Effectiveness of Hydroxyurea in the Management of the Painful Crisis in Sickle Cell Anemia
© 2021 Shanmugakumar S D, Parameshwar P, Mahendar K, Afreen Banu, Sobia Kaleem, et al, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Sickle cell syndromes are inbred disorders distinguished by the presence of blade (Sickle) hemoglobin in red blood corpuscles. The sickling of red cells in patients with SCA is caused by the polymerization of molecules of non-oxygenated hemoglobin S (α2ß2s) into rigid rod-like polymers. In the open-label study of hydroxyurea therapy, the synthesis of embryo hemoglobin increased in most patients with SCA resulting in a humane myelotoxicity and rise in painful crises. By suppressing the sickling process, increased levels of embryo hemoglobin sinew the less frequency of painful crises.
Methods: In prospective observational learning, we assessed the efficiency of hydroxyurea in decreasing the recurrence of painful episodes associated with the reduction in the prevalence of blood transfusion in children and adults with a history of complications. The trial was aborted after a mean follow-up of 4 months.
Results: Among 80 patients investigated, 41 patients were men and 39 patients were women. The prospective observational study in the department of pediatrics, Prathima Institute of Medical Sciences, Nagunoor, Karimnagar, T.S. The 80 patients designate to hydroxyurea treatment, out of which 64 patients were taking hydroxyurea in the duration of 0-5 yrs; 12 patients were taking hydroxyurea for a span of 6-10 yrs old; 4 patients were taking hydroxyurea for the period of 11-15 yrs. No patients were managed with hydroxyurea with the greater of 16 yrs. The dose is administered in the range of 15-35mg/kg per day. The serum ferritin levels, hemoglobin, direct, and total bilirubin levels were investigated for three follow up. It was found that F – statistical value -0.41467, P-value -0.66104 & right- tailed p-value (Z>z) -0.6554217 for Serum ferritin; F- statistical value – 0.9209, P-value - 0.3996 & right-tailed p-value (Z>z)-0.3594236 for haemoglobin ; F-statistical value - 0.92093P-value-0.39957 & right-tailed p-value (Z>z)-0.3993615 for total bilirubin ; F-Statistical value -1.11703,P-value – 0.32897 & right-tailed p-value ; P (Z>z) – 0.3993615 for direct bilirubin respectively. During the treatment with hydroxyurea, it was observed that none of the patients were reported the unwanted effects with hydroxyurea.
Conclusion: Hydroxyurea therapy can ameliorate the clinical course of SCA in children and adults with three or more painful crises. Maximally tolerated doses of hydroxyurea may not be necessary to achieve a therapeutic effect. The long-term safety of hydroxyurea in patients with SCA is uncertain.
Introduction
The erythrocytes of unequivocal individuals will undertake commutative changes in morphology in retaliation to the fragmentary pressure of oxygen. When the oxygen pressure is let down, these cells will alter their forms from biconcave disk to crescent, holly wreath, and other forms. This phenomenon is denoted as sickling. Nearly 8 % of the global population possess this characteristic; usually, they exhibit no pathological consequences ascribable to it. These people are said to have sicklemia or SCA [1]. In sickle cell hemoglobin, a sole point mutation in the DNA codon for glutamic acid pops up to result in a base triplet that is interpreted into a message RNA (mRNA), which cipher for valine. The series of the standard mRNA has been recently elucidated and indicates that normal glutamate is coded at the ribonucleotide triplet of guanosine - adenosine – guanosine. A single base conversion of adenosine to uridine ought to build the messenger codon guanosine-uridine - guanosine ( GUG) that codes for valine. It is presumed that the globin chain is synthesized with the utilization of the atypical m RNA template and the atypical globin chain then complexes with alpha chains to obtain the sickle hemoglobin tetramer ( 2, 2) which is called sickle cell hemoglobin [2]. Sickle hemoglobin has the singular property of forming polymers when oxygenated. On deoxygenation, over there is a moratorium varying from milliseconds to seconds in advance of hemoglobin delta polymers can be remarked, after which they assemble rapidly. The scope of the moratorium varies among erythrocytes containing hemoglobins and is exquisitely dependent on the engrossment of hemoglobins. Sickle cell trait is amiable, due to the cellular concentration of hemoglobin S is too low for polymerization to occur under most conditions. Among hemolytic anemias, the vaso- occlusive features of sickle cell disease are eccentric. Deoxygenation of sickle cells induces potassium efflux which increases cell density and the tendency of hemoglobin S to polymerize [3]. About 60% of patients will have an interlude of severe pain.
The differences exist as one of the manifestations of heterogeneity of this disease; which complicates the possibility of treatment. Patients with severe pain should be given an opiate parenterally at frequent, fixed intervals not as needed until the pain has fallen off at which time the dose of the opiate can be tapered and then aborted and oral analgesic therapy can be initiated. Pain should be monitored frequently with the aid of pain measurement scales to lead the severity of treatment. Most patients who possess episodes of acute pain are not drug addicts or drug wanters. Transfusions are not required for the usual anemia or events of pain associated with sickle cell disease. Desperate replacement of blood is often required for sudden severe anemia occurring in children when blood is sequestered in an expanded spleen. Recent clinical trials have evaluated the efficacy of the transfusions in patients with some obstacles of sickle cell disease. Repeated transfusions reduce the probability of recurrent stroke in children with sickle cell anemia [4].
Overall 30 years of research documenting its recognized laboratory and clinical benefits for sickle cell Anemia. The channels by which hydroxyl urea induces HbF and ameliorates the pathophysiology of sickle cell anemia remain incompletely understood. The primary advantage of hydroxyurea for sickle cell anemia relates to its ability to rise HbF [5].
The main objective of this work is to assess the effectiveness, efficacy, and safety of hydroxyurea in sickle cell anemia by observing the total number of transfusions before and after initiation of hydroxyl urea and adverse effects associated with hydroxyurea. Further to document the demographic features of the patient’s family medical history, age of diagnosis, the total number of transfusions, adverse effects associated with hydroxyl urea. Laboratory investigations like the serum, ferritin, hemoglobin, direct and indirect bilirubin. To evaluate the complications of sickle cell anemia.
Materials and Methods Study design
A retrospective observational study was carried out in the pediatrics department of the transfusion ward on those diagnosed with sickle cell anemia and who were prescribed hydroxyl urea.
Calculation of sample size
Based on the WHO guidelines, the sample size calculation in health sciences [6]. A total of 80 patients from various regions of Telangana are admitted to the inpatient department of the transfusion ward. The patients were asked different questions regarding their family history; medical history and medication history and health condition.
Study criteria Inclusion criteria
All the patients who were admitted to the pediatrics ward as in patients with sickle cell anemia had two or more painful crises. Screening of sickle cell anemia patients on medication with hydroxyl urea.
Exclusion criteria
Children with less nutrition: Malignancy or other chronic conditions that could stimulate hydroxyl urea toxicity: Beta – Thalassemia patients.
Sampling technique and data collection
The informed consent was collected from all the patients before conducting the study. A well–designed patient data collection form was developed and used for this study. A proforma was designed and pretested to be used for enrollment patient’s specific information. The study was performed by utilizing the patient case sheets, laboratory data, direct – communication with patients and their caregivers. During the study period (late 2020 through early 2021) scheduled visits to the wards and outpatient department by the study field teams.
Data were collected using a structured questionnaire. It consists of details of the patient’s past medical history: family medical history: laboratory data that induces a complete blood picture; serum ferritin: medication history. The patient was reviewed daily who was admitted to the transfusion ward. The information data was collected in a properly designed facts collection form. All the patients were advised with hydroxyurea with a maximum dose of 35mg/kg to reduce the frequency of blood transfusion and to lower the frequency of painful crises.
Statistical analysis of data
Descriptive statistics are utilized to report the variables of interest. Categorical variables are reported as absolute and relative frequencies. Continuous variables are reported as means and standard deviation or median or interquartile range. Normally distributed data were analyzed using ANOVA analysis.
Results
The present study includes 80 patients attending the department of pediatrics, prathima institute of medical sciences, Nagnoor, Karimnagar. Telangana state. The study showed that 36 patients were in the age group of 11-15 yrs. 31 patients were in the age group of 6-10 yrs. 11 patients were in the age group of 16-20 yrs. 2 persons were in the age group of 20-25 yrs.
Table 1 shows the distribution of sickle cell anemia patients based on their age and gender. Persons aged less than 6 yrs old were not taken for study purposes. It was also observed that the risk of sickle cell anemia was greater among male children compare to females. Based on their family history it was observed that with a specific gender, there is no difference in the transfer of genes but the trait genes are responsible for the transfer of sickle cell anemia. Table 2 shows the distribution of sickle cell anemia patients based on immunization. Out of 80 sickle cell anemia patients scrutinized; 53.75 % of patients were vaccinated with meningococcal vaccine.
53.75 % were vaccinated with pneumococcal vaccine and 48 % were vaccinated with Haemophilus Influenzae type B vaccine.
Table 1: Distribution of SCA patients based on their age and gender
|
Age in years |
Male |
Female |
Total |
|
1-5 |
0 |
0 |
0 |
|
6-10 |
14 |
17 |
31 |
|
11-15 |
25 |
11 |
36 |
|
16-20 |
2 |
9 |
11 |
|
20-25 |
0 |
2 |
2 |
|
Total |
41 |
39 |
80 |
Table 2: Distribution of SCA patients based on immunization
|
Father |
Mother |
Parents |
Siblings |
|
36 |
29 |
10 |
5 |
Table 3 shows the distribution of sickle cell anemia patients based on pain scale readings. Out of 80 sickle cell anemia persons investigated, 4 patients had extremely painful conditions which are unbearable ( pain scale 10). 30 patients had severe painful crises (pain scale 8). 22 patients had moderate pain crises (pain scale 6) . 18 patients had tolerated painful crises (pain scale 4). 6 patients had mild pain (pain scale 2). None of the patients were been without pain episodes.
Table 3: Distribution of SCA patients based on their pain scale readings
|
Pain Scale readings |
No: of Patients |
|
0 |
0 |
|
2 |
6 |
|
4 |
18 |
|
6 |
22 |
|
8 |
30 |
|
10 |
4 |
Table 4 shows the distribution of sickle cell anemia patients based on their type of DNA. Out of 80 patients, 24 (30%) patients were homozygous and 56 (70%) patients were heterozygous.
Table: 4 Distribution of SCA patients based on their type of DNA
|
DNA Type |
No: of patients |
Percentage |
|
Homozygous |
24 |
30% |
|
Heterozygous |
56 |
70% |
Table 5 shows the distribution of sickle cell anemia patients based on their complications history. Out of 80 sickle cell anemia patients surveyed based on their former complications; the major part of the patients (36 ) had suffered from both hepatomegaly and splenomegaly; 28 patients were suffering from splenomegaly; On the contrary, 12 patients were suffering from hepatomegaly; only 5 patients were having evidence of infection with hepatitis –C. A very few patients (4) were free from complications.
Table 5: Distribution of SCA patients based on their complications history
|
Hepatomegaly |
Splenomegaly |
Both – Hepatomegaly & splenomegaly |
Nil |
Hepatitis - C |
|
12 |
28 |
36 |
4 |
5 |
Table 6 shows the distribution of patients based on their age in which sickle cell anemia was diagnosed. Out of 80 sickle cell anemia patients explored based on the age in which the disease was diagnosed, it was identified that major 66 patients were from 0-5 yrs old infantry. 9 patients have evidenced sickle cell anemia in the range of 6-10 yrs old toddler. 3 patients were found to possess sickle cell anemia in the age of 11-15 yrs old adolescent.2 patients have been diagnosed with sickle cell anemia in the teenage of 16-20 yrs old.
Table 6: Distribution of patients based on their age SCA was diagnosed
|
AGE |
No: of Patients diagnosed with SCA |
|
0-5 yrs |
66 |
|
6-10 yrs |
9 |
|
11-15 yrs |
3 |
|
16-20 yrs |
2 |
|
Total |
80 |
Table 7 exhibits the distribution of sickle cell anemia patients based on their age at which hydroxy urea was commenced. Out of 80 patients with sickle cell anemia disease and the age at which hydroxy urea was started. It was identified as 44 patients were possessing toddler age of 6-10 yrs. 21 patients were 0-5 yrs old infantry. 13 patients were possessing adolescent category of 11- 15 yrs old. 2 patients were categorized under teenagers age about 16-20 yrs respectively.
Table 7: Distribution of SCA patients based on their age at which hydroxyurea was commenced
|
Age |
No: of Patients at which hydroxyurea was started |
|
0-5 yrs |
21 |
|
6-10 yrs |
44 |
|
11-15 yrs |
13 |
|
16-20 yrs |
2 |
|
Total |
80 |
Table 8 showed the duration of the treatment of hydroxy urea in sickle cell anemia patients. Out of 80 sickle cell anemia patients studied for the treatment of hydroxy urea, it was identified that 64 patients were under the category of infantry in the range of 0-5 yrs old. 12 patients belong to a class of toddler age of 6-10 yrs old. 4 patients be in the hands of adolescent age of 11-15 yrs old. None of the teenagers were treated with hydroxyl urea.Table 9 showed the distribution of treatment of dose of hydroxyurea to sickle cell anemia. All 80 patients received 500mg of hydroxyurea for the treatment of sickle cell anemia.
Table 8: Duration of the treatment of hydroxyurea for SCA patients
|
Duration |
No: of Patients |
|
0-5 yrs old |
64 |
|
6-10 yrs old |
12 |
|
11-15 yrs old |
4 |
|
16-20 yrs old |
0 |
|
Total |
80 |
Table 9: Treatment of dose of the hydroxyurea to sickle cell anemia
|
Total Number of SCA patients studied |
Dose of hydroxyurea |
Percentage of treatment |
|
80 |
500mg |
100% |
Statistical Analysis
Descriptive statistics are utilized to report the variables of interest. Categorical variables are observed as absolute and relative frequencies. Normally distributed data were analyzed using ANOVA analysis. Univariate ANOVA is reviewed from a user –point of view with emphasis on understanding the model building and the predictions underlying the method. ANOVA analyses were performed for serum ferritin levels; hemoglobin levels: total bilirubin: direct bilirubin levels respectively (Table 10-12; Figure:1-4).
Table 10: Analysis of Variance results of serum ferritin levels in sickle cell anemia patients
F-Statistical Value = 0.41467 P-Value = 0.66104
|
Data summary |
|||||
|
Groups |
N |
Mean |
Standard deviation |
Standard error |
|
|
Group-1 (Serum Ferritin V1) |
80 |
2605.75 |
2770.6582 |
253.8673 |
|
|
Group-2 ( Serum Ferritin V2) |
80 |
2813.25 |
2872.4771 |
321.1527 |
|
|
Group-3 ( Serum Ferritin V3) |
80 |
243.65 |
2681.8589 |
299.8409 |
|
|
ANOVA SUMMARY |
|||||
|
Source |
Degrees of freedom DF |
Sum of squares SS |
Mean Square MS |
F-Stat |
P-Value |
|
Between groups |
2 |
5694632.6333 |
2847316.3167 |
0.4147 |
0.661 |
|
Within groups |
237 |
1627351060.3501 |
6866460.1703 |
|
|
|
Total |
239 |
163304562.9834 |
|
|
|
Table 11: Analysis of Variance results of hemoglobin levels in sickle cell anemia patients
F-Statistical Value = 0.92093 P-Value = 0.39957
|
Data summary |
|||||
|
Groups |
N |
Mean |
Standard deviation |
Standard error |
|
|
Group-1 (Hemoglobin V1) |
80 |
2.6825 |
2.8741 |
0.3213 |
|
|
Group-2 (Hemoglobin V2) |
80 |
2.3375 |
2.0926 |
0.234 |
|
|
Group-3 (Hemoglobin V3) |
80 |
2.2325 |
1.3434 |
0.1502 |
|
|
ANOVA |
|||||
|
Source |
Degrees of freedom DF |
Sum of squares SS |
Mean Square MS |
F-Stat |
P-Value |
|
Between groups |
2 |
8.868 |
4.434 |
0.9209 |
0.3996 |
|
Within groups |
237 |
1141.0878 |
4.8147 |
|
|
|
Total |
239 |
1149.9558 |
|
|
|
Table 12: Analysis of Variance results of total bilirubin levels in sickle cell anemia patients
|
Data summary |
|||||
|
Groups |
N |
Mean |
Standard deviation |
Standard error |
|
|
Group-1 (Total Bilirubin V1) |
80 |
2.6825 |
2.8741 |
0.3213 |
|
|
Group-2 (Total Bilirubin V2) |
80 |
2.3375 |
2.0926 |
0.234 |
|
|
Group-3 ( Serum Ferritin V3) |
80 |
2.2325 |
1.3434 |
0.1502 |
|
|
ANOVA SUMMARY |
|||||
|
Source |
Degrees of freedom DF |
Sum of squares SS |
Mean Square MS |
F-Stat |
P-Value |
|
Between groups |
2 |
8.868 |
4.434 |
0.9209 |
0.3996 |
|
Within groups |
237 |
1141.0878 |
4.8147 |
|
|
|
Total |
239 |
1149.9558 |
|
|
|
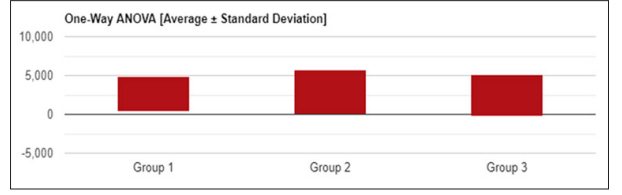
Figure 1: Univariate ANOVA (Average +/-Standard deviation) of Serum Ferritin in SCA patients
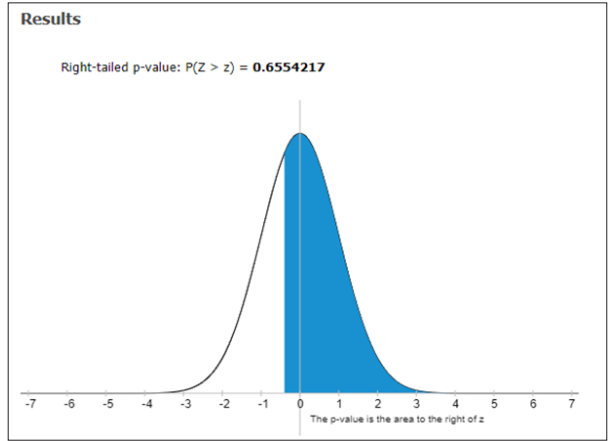
Figure 2: Right –tailed P-value P (Z>z) of serum ferritin in SCA patients
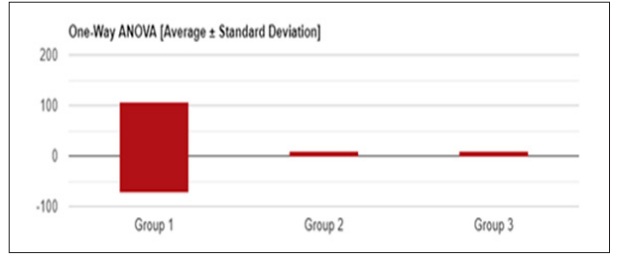
Figure 3: Univariate ANOVA (Average +/-Standard deviation) of hemoglobin in SCA patients
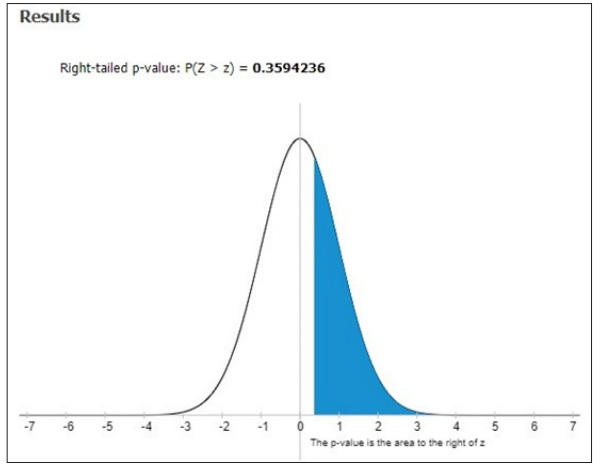
Figure 4: Right–tailed P-value P (Z>z) of hemoglobin in SCA patients
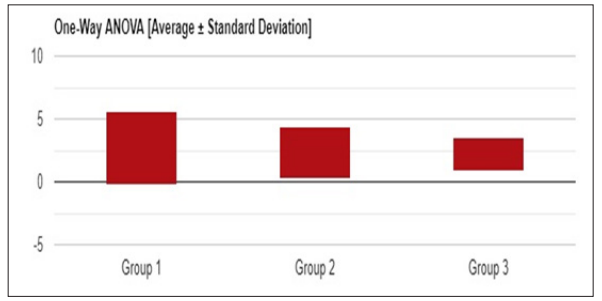
Figure 5: Univariate ANOVA (Average +/-Standard deviation) of Total Bilirubin in SCA patients
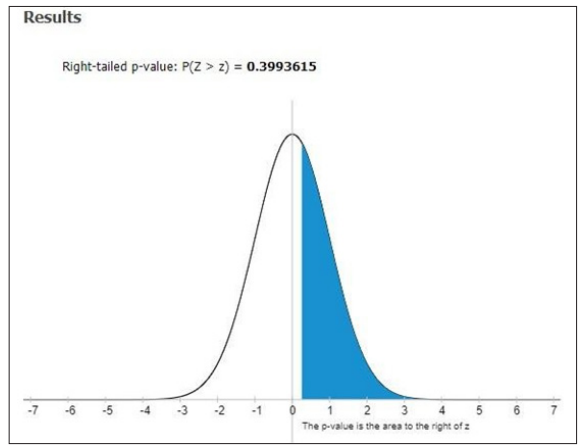
Figure 6: Right–tailed P-value P (Z>z) of Total Bilirubin in SCA patients
Discussion
The overall investigated patients aught were possessed with sickle cell anemia were healed with daily doses of hydroxyl urea to assess their pharmacokinetic behavior. Overall 80 sickle cell anemia patients were studied for the utilization of hydroxyl urea in the dose range of 10- 500mg/kg based on their body weight. It was found that 2.6% to 27.8% with an elevated level in hemoglobin F on an average of 1.25g/dL. Our statistical analysis showed that F=0.92093 and P-Value = 0.39957 ; DF – 239 ; SS – 1149.9558 ; F- Stat – 0.9209 ; P-Value – 0.3996; Right – tailed P-value – 0.3594236 respectively. In an aspect of demographic details out of 80 patients examined, the majority belongs to the age group of 11-15 yrs. 36 patients ( M-25, F-11) followed by 31 patients (M-14, F-17) of the age group of 6-10 yrs. 11 patients ( M-2: F-9) were in the age group of 16-20 yrs. Only 2 patients (M-0, F-2) of the age group of 20-25 yrs. Further in our learning, it was identified that 5 siblings were obtained sickle cell anemia disease from parents, father or mother traits respectively. The distribution based on immunization revealed that 43 patients were vaccinated with the meningococcal vaccine, 43 patients were vaccinated with pneumococcal vaccine and 39 patients were immunized with Haemophilus influenza vaccine.
Based on the studies about the pain scale readings, it was identified that out of 80 patients investigated, 4 patients were at the pain scale readings at 10; 30 patients had been at the pain scale readings at 8; 22 patients had moderate pain scale as 6; 18 patients had tolerated painful episodes 4; 6 patients had mild pain of pain scale 2. It was observed that none of the sickle cell anemia patients were been without pain episodes.
In our investigation, out of 80 patients, the major contributes heterozygous i.e. 70 %. While differentiating to homozygous 30 %. Hence it was determined that heterozygous patients were less chance compared to the homozygous aspect of the slighter elevation of Sr. Ferritin levels. The need for blood transfusion was less. ANOVA studies for blood transfusion were less. ANOVA studies for Sr. Ferritin in our study of 80 patients were revealed that F= 0.414167, P-Value = 0.66104: Degrees of freedom DF:239. SS value: 163304562.9834 : F- Stat -0.4147 ; P-Value – 0.661 :
Right –tailed P-Value – 0.6554217 respectively. In our distribution study based on complications history, it was concluded that out of 80 sickle cell anemia patients examined; 12 patients were suffering from hepatomegaly; 28 patients were suffering from splenomegaly; 36 patients were put up with hepatomegaly & splenomegaly; 4 patients did not have any history of complications. 5 patients were enduring hepatitis –C respectively.
In our study, out of 80 patients explored based on the age at which the sickle cell anemia disease was diagnosed. It was identified that the majority of 66 patients were from 0-5 yrs old infantry. 9 patients were evidence sickle cell anemia in the range of 6-10 years toddler. 3 patients were found to possess sickle cell anemia to the age of 11-15 yrs old adolescent.2 patients were diagnosed with sickle cell anemia in the teenage of 16-20yrs old. Further, out of 80 patients focused on the treatment of hydroxyl urea in painful crisis in sickle cell anemia, it was concluded that 64 patients were under the category of infantry in the range of 0-5 yrs old. 12 patients belong to the class toddler age of 6-10 yrs old: 4 patients to be in hands of adolescent age of 11-15 yrs old. None of the teenagers were treated with hydroxyl urea. Statistical analysis of total bilirubin and direct bilirubin in sickle cell anemia patients. ANOVA studies revealed that F- Value -0.92093 & 1.11703 ; P-Value – 0.39957 & 0.32897 ; DF -239 ; SS -1149.9558 & 45.648 ; F-Stat – 0.9209 & 1.117 ; Right- tailed P-value –0.3993615 respectively. Hydroxyurea is the first clinically acceptable drug shown to abort a painful crisis in children and adults with sickle cell anemia. Our facts support the use of hydroxyl urea therapy for the prevention of painful situations in adults and children who can follow directions about dosages and evaluating the obscure long-term risks of such treatment.
Conclusion
The outcome of learning ends up that patients taking hydroxyl urea for frequent painful sickle cell anemia episodes appear to have less impermanence after 9 years of follow-up. Survival was related to hemoglobin F levels; serum ferritin levels; Total bilirubin & direct bilirubin levels and related vaso-occlusive events periodically. Further, it came to know that hydroxyl urea increases hemoglobin F concentrations and diminishes vaso- occlusive complications in children and adults with moderate to severe sickle cell anemia and these effects were associated with less death rate. After up to 9 years of follow-up, unpredicted serious side effects of this treatment were not found. Whether hydroxyurea should be provided to patients with sickle cell anemia remains to be untapped. Hence we conclude that the underlying phenomenon of disease severity remnant critical to identify the prognosis of adult sickle cell anemia, but hydroxyurea will be able to reduce the severity of the sickle cell anemia disease.
Author’s Contributions
All contributing authors have agreed to the submission of this manuscript for publication. SPSS package software was utilized for the statistical analysis of data. All authors have read and approved the final version of this manuscript.
Funding
The study was self-funded
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors acknowledge the full cooperation of the staff of the department of pediatrics, Prathima Institute of Medical Sciences, Karimnagar in the present study. Further authors were thankful to Sri J.Sagar rao and Sri J.Sumith sai, Chairman and Secretary & Correspondent of Jyothishmathi group of institutions, Karimnagar for their support and encouragement.
References
- Linus pauling, Harvey A Itano, Singer SJ, Ibert C wells (1948) Sickle cell Anemia, a molecular disease science 110: 543-548.
- Marotta CA, Forget BG, Cohensolal M, J T Wilson, S M Weissman (1977) Human β –globin messanger RNA I. Nucleotide sequences derived from complementary RNA. J.Biol.Chem 252: 5019-5031.
- Joiner CH (1993) Cation transport and volume regulation in sickle cell red blood Am. J. Physiol. Cell Physiol 264: C 251-C270.
- Platt OS, Brambilla DJ, Rose (1994) Mortality in sickle cell disease: Life expectancy and risk factors for early death. N.Engl.J.Med 330:1639-1644.
- Jurrien Dean, Alan N Schechter (1978) Sickle cell – Anemia: Molecular and cellular bases of therapeutic approaches. N.Engl. J. Med 17: 496-508.
- Lwanga SK, Lemeshow S (1991) World health. Sample size determination in health studies. A Pratical Manual Organization 122-126.