Author(s): <p>Alemu Tsega* and Destaw Mullualem</p>
Hemophilia is the X-linked inherited bleeding illness caused by genetic abnormalities in the F8 (hemophilia A) or F9 (hemophilia B) genes. Hemophiliacs experience bleeding episodes into the joints and soft tissues, which can be managed with self-infusion of concentrations of factor VIII or IX. Because hemophilia is a monogenic illness with easily measured outcomes (factor levels and bleed rates), it makes it a desirable target for gene therapy. One of the most promising vectors for hemophilia gene therapy is lentiviral or adeno-associated virus (AAV)-based. Recombinant adeno-associated viral (AAV) vectors have been used successfully in all preclinical and clinical trials to far for factor VIII or IX hepatocyte transduction. These vectors were used to repair the bleeding phenotype in preclinical animal models of hemophilia A and B. Clinical translation to individuals with severe hemophilic phenotype was spurred by some of these encouraging preclinical results. This represents a significant advancement in the field because these patients undergoing gene therapy with AAV vectors demonstrated long-term expression of therapeutic FIX levels. With gene therapy, hemophilia patients can have a single treatment and maintain their factor levels for years or decades rather than relying on a steady supply of medication and frequent delivery at short intervals. Over the past ten years, significant progress has been achieved in the development of gene therapy for hemophilia. The phase III trial stage of adeno-associated virus (AAV) vector-mediated gene transfer in hemophilia A and B has begun. Clinical trials are currently evaluating factor VIII (FVIII) or factor IX (FIX) gene editing technologies or lentiviral vector gene transfer. It is anticipated that major markets will authorize and distribute the first gene therapy drugs, FVIII and FIX, in the near future.
Hemophilia is a genetic disorder characterized by the deficiency of a plasma protein needed for normal blood clotting. The two most common forms of hemophilia are hemophilia A and B; both are classically transmitted in an X-linked recessive pat-tern, with one third of cases due to de novo somatic mutation [1]. X-linked bleeding illness called hemophilia affects about 1 in every 5,000 male babies born worldwide. It is brought on by mutations in blood dressing factor VIII (FVIII, hemophilia A) or factor IX (FIX, hemophilia B) [2]. The X-linked genetic coagulation disorder hemophilia A is brought on by a lack of functioning clotting factor VIII (FVIII). Based on the remaining FVIII activity, the disease is categorized as mild (>5–<40 IU/dl), moderate (<1 IU/ dl), or severe (<1 IU/dl) [3]. The X-linked hereditary bleeding disorder hemophilia B is characterized by spontaneous or induced frequently uncontrollable bleeding into the muscles, joints, and other soft tissues. A mutation in the gene that produces factor IX is the cause of the illness. It impacts 1 in 25 male births [4].
A coagulation factor deficit brought on by low or absent amounts of factor IX is known as hemophilia B. Reduced levels or a lack of plasma FIX, which cleaves and activates FX within coagulation cascade, causes symptoms of recurrent prolonged bleeding. FIX is a plasma protease that is dependent on vitamin K and functions within the intrinsic blood coagulation system [5]. The initial amount of clotting factor activity is used to classify the severity of hemophilia. The activity levels of Factor VIII are expressed in units, where 1 U ml–1 represents 100% of the factor present in 1 milliliter of normal plasma. Typically, normal plasma activity levels fall between 0.5 and 1.5 U ml–1 [6]. Before the 1970s, there was no effective therapy for severe hemophilia, and most individuals only lived until childhood or early adulthood owing to bleeding in key organs (with intracranial bleeds being particularly prevalent). individuals also lived with significant physical limitations [7]. Before clotting factor, preparations were developed, people with hemophilia had a mean life expectancy of less than thirty years [8].
Many people have the misconception that bleeding disorders only affect men. They also think that women ‘simply’ carry the mutated gene and can pass it on to their sons. However, women may experience severe symptoms. Affected males experience symptoms. They pass the influenced X chromosome to their daughters instead of their sons. Females carrying the influenced X chromosome typically do not exhibit symptoms. However, they have a 50% chance of passing the influenced gene to their offspring [9]. Over the past few decades, hemophilia treatment has seen significant progress. The aim of treatment changed from managing bleeding to preventing bleeding, with prophylactic factor concentrate replacement replacing on-demand bleeding. Furthermore, the goal of eliminating chronic joint disease and attaining a nearly normal life expectancy developed along with prevention [3]. Hemophilia is an X-linked bleeding diathesis resulting from a deficiency of blood coagulation factor VIII (F. VIII) (hemophilia A) or factor IX (F.IX) (hemophilia B) [10]. Clinically, the disease is characterized by frequent spontaneous bleeding episodes, mostly into joints or soft tissues. Bleeding can also occur into other critical closed spaces, such as the intracranial space or the retroperitoneal space, where it can be rapidly fatal. Hemophilia A occurs in ’1 in 5000 male births; hemophilia B is less common, occurring in ’1 in 30 000 births [11].
One-time treatment that has the potential to produce prolonged coagulation factor expression even within the normal range is gene therapy, which has emerged as a paradigm-shifting therapeutic approach. It has been explored for many years, but only in the past ten years have there been an increasing number of various clinical trials and significant therapeutic outcomes; it feels both fresh and old at the same time. The European Medicine Association (EMA) has only licensed one gene therapy product thus far, but the hemophilia community is still striving to learn more and comprehend this game-changing medicine [12].
The mode of transmission for HA and HB is X linked recessive. The X chromosomes long arm contains the HA and HB genes, which are found in bands Xq28 and Xq27, respectively. Therefore, the son of a female carrier has a 50% risk of inheriting the condition. A man who has hemophilia won’t pass the illness on to his son, but all of his daughters will carry it. Although female carriers of hemophilia typically do not exhibit symptoms, they may have clotting factor levels that are lower than normal [13].
The 186 kb factor VIII gene is a huge and complex gene. Numerous genetic flaws, such as a wide range of point mutations, gene deletions, abnormalities in stop codons, frame shift mutations, and inversion mutations, can cause hypoplastic behavior (HA). These insights aid in the identification of carriers through DNA analysis and the repair of gene defects to stop the generation of inhibitors [14].
A uncommon X-linked congenital condition known as hemophilia, which results in a lack of clotting proteins FVIII or FIX, causes “prolonged, spontaneous, potentially life-threatening bleeding [15]. Recurrent bleeding events can cause long-term, incapacitating consequences; they occur most often in the muscles and joints. As previously stated, hemophilia cannot be cured, but it can be controlled by treating bleeding episodes as soon as they occur using infusions of FVIII for hemophilia A or FIX for hemophilia B.Desmopressin or anti fibrinolytics may be administered to hemophilic patients in specific circumstances [15].
Factor VIII and IX, which are necessary for the coagulation cascade to propagate for thrombin generation, are deficient in hemophilia A and B, two X-linked recessive bleeding disorders. Together, they account for around 0.01% of all severe inherited bleeding diseases, making them the most prevalent [16]. Hemophilia vary in their clinical presentation; severe disease (factor level <0.01 IU/mL) is characterized by spontaneous bleeding into muscles and joints; moderate hemophilia (factor level 0.01–0.05 IU/mL) and mild hemophilia (factor level >0.05 IU/mL) are characterized by excessive bleeding following trauma or surgery(14). Since de novo mutations produce about one-third of hemophilia cases, these individuals do not have a family history of the condition [17]. Hemophilia is mostly a male trait due to X-linked heredity; however, low levels of FVIII or FIX and a bleeding diathesis due to skewed X inactivation can also affect female carriers.
Rarely, homozygous or compound heterozygous inheritance of F8 or F9 mutations can also cause hemophilia in females. In addition, Turner’s syndrome, Testicular Feminization Syndrome, and loss of heterozygosis must be taken into account [18]. There are current studies examining the pathophysiology of irregular bleeding in hemophilia carriers, which is not rare. Finding the underlying F8 or F9 mutation is crucial for carrier and prenatal diagnosis, as well as for predicting the risk of inhibitor formation or anaphylactic reaction after infused replacement therapy, even though the diagnosis of hemophilia is usually based on the much faster and more readily accessible clotting factor levels rather than genetic analysis (with the exception of prenatal cases) [19].
The various cutting-edge therapies, including tissue engineering, gene therapy, and cell therapy, along with the more recent development of induced pluripotent stem cell (iPSC) technology, may in the future provide countless clinical applications for the treatment of certain monogenic diseases, including hemophilia. Although it is true that hemophilia can be effectively treated with advanced therapy protocols due to its monogenic nature and the fact that a slight increase in coagulation factor levels can change a severe phenotype into a moderate one, research in this area is still in its infancy, and significant work needs to be done before such therapies are widely accessible to patients, especially in terms of safety Particularly in this group of patients who appear with certain clinical characteristics that are almost certainly the effect of their prior and current treatment, safety considerations will need to be handled very seriously. These features include the presence of inhibitors or the propensity to acquire them, the patients’ unique immunological condition, and the existence of co-infections with viruses (HIV/HCV). For these reasons, prudence is crucial to prevent inflating expectations in both doctors and patients, even though optimism is undoubtedly warranted [20].
Hemophilia is completely cured with gene therapy. Adeno- associated viral vector gene therapy for hemophilia B has shown encouraging outcomes in certain investigations. It produces a transient remission, and more research is necessary to find a long-term solution Some constraints that restrict its application include toxicity, safety concerns, humoral and cellular immune response, and these need to be addressed in the future [21,22]. In hemophilia A, antibodies against recombinant proteins develop. Giving newborns an adenoviral vector expressing factor VIII can avoid this because of their underdeveloped immune systems [23,24]. Hemophilia is most likely the hereditary condition with the combination of characteristics that increases the likelihood of a positive outcome from gene replacement treatment. These disorders’ clinical symptoms are solely caused by the absence of a single gene product, which is present in trace amounts in the population. Strictly controlled gene expression regulation is not necessary; in extreme situations, a little increase in the protein’s plasma level significantly improves symptoms. There are models of animals available. Coagulation factors can be produced by a wide variety of cell types, and the location of synthesis is not essential to function [25].
Decades of preclinical and clinical work have resulted in the current successful phase I/II gene therapy trials for hemophilia. All published and currently open studies use adeno-associated viral (AAV) vectors [26]. Gene therapy involves introducing genetically modified cells into an organism to enable them to create a functional protein, while cell treatment involves introducing living cells into an organism to facilitate tissue repair or restore a function that is impaired. Given that stem cells have the endless potential to self- renew and specialize into a variety of distinct cell lines, both tactics rely on their employment. For its clinical application, each of these prerequisites must be met [27]. Preclinical and clinical studies in the domains of gene therapy (using viral and non-viral vectors) and cell therapy (using various target cell types) have yielded the most notable advances in the disciplines of advanced therapeutics and hemophilia (Table 1). Therefore, hemophilia shows itself to be a condition that responds well to gene therapy by means of lent viral and adeno-associated vectors used in adult stem cells and autologous fibroblasts, platelets or hematopoietic stem cells; and of the transfer of nonviral vectors and repair of mutations by chimeric oligonucleotides [13,28]. In terms of immune-mediated transgene rejection (factor VIII or IX expression), the majority of published studies have not documented any adverse events resulting from the application of such strategies in clinical trials. However, their success is being hindered by factors like innate cellular T cell toxicity to adeno-associated capsid protein and the low efficacy obtained by non-viral vectors [29]. The development of inhibitory alloantibodies against FVIII or FIX has proven to be the most difficult side effect of treatment. These inhibitors are developed in about 25–30% of cases of severe hemophilia. Reduce the efficacy of replacement medicines in hemophilia A patients and in just 3-5% of hemophilia B patients, restrict patient access to a safe and effective standard of care, and put patients at higher risk of morbidity and death [4].
An effective prophylactic that stops bleeding in joints in both children and adults with hemophilia is essential for a positive long-term result for people with the disorder. Effective prophylaxis necessitates considering the amount of permissible bleeding, particularly joint bleeds, as well as the available resources (clotting factor concentrate, trough levels), the bleeding trigger (activity levels, chronic synovitis, pre-existing arthropathy), and finally, the accessible resources [30]. Adeno-associated virus (AAV) was the first vector linked to curative gene transfer in animal models of hemophilia; at this point, AAV vectors are the only ones that can be utilized to provide hemophilia patients therapeutic doses of FVIII and IX.66–70 Ten individuals with severe HB at the Royal Free Hospital in London, UK, participated in the initial trial in the past. These patients received single, escalating doses of an AAV8 vector, with some of them undergoing a current follow-up of nine to ten years [31].
A single gene deficiency causes hemophilia, and factor levels in plasma can be used to measure gene expression with ease. Even little expression of FVIII or FIX significantly improves the bleeding phenotype. Low expression of FVIII was found in the first investigations employing an ex vivo method of gene therapy via gene transfer in autologous fibroblasts or hematopoietic stem cells. Thirteen years ago, individuals with hemophilia B were injected intramuscularly with recombinant AAV (rAAV)-FIX as part of an Adeno-associated virus (AAV)-based gene therapy. Though expression in muscle biopsies persisted for almost three years, the technique was safe, and there was only a little increase in plasma FIX as a result [32]. For transgene delivery, alternative viral and non-viral-based vectors are needed due to pre-existing AAV humoral immunity and the possible gradual loss of vector transduction. Lentiviral vectors, which have a lower incidence of pre-existing humoral immunity and a larger packaging capacity than AAV vectors, have been the main approach employed in hemophilia research. A review of pre-clinical research utilizing blood outgrowth endothe- lial cells or in-vivo or ex-vivo stem cell transduction (either hematopoietic or induced pluripotent) has been published [26].
|
Inserted gene |
Name |
Campany |
phase |
Period |
Vector used |
Identification code |
|
F8 |
Valoctogene roxaparvovec(AAV5- Hfvii-SQ) |
BOM-270:BioMarin |
III |
2015- 2024 |
AAV5 |
NCT02576795 |
|
SPK-8011 |
Spark Therapuetics |
VIIa |
2017- 2020 |
rAAV |
NCT033003533 |
|
|
SB-525 |
Sangamo and pfizer |
VIIa |
2017-2024 |
AAV2/6 |
NCT03061201 |
|
|
SHP654 |
Shire |
VIIa |
2018-2024 |
AAV8 |
NCT03370172 |
|
|
F9 |
SPK90001 |
Spark Therapuetics and pfizer |
VII |
2017-2026 |
AAV |
NCT03307980 |
|
AMT060 |
UniQure |
VII |
2015-2021 |
AAV5 |
NCT02396342 |
|
|
AMT061 |
UniQure |
III |
2018-2024 |
AAV5 |
NCT03569891 |
|
|
Sc AAV2/8 LP1- HFIXco |
UCL and SJCRH |
VII |
2010-2032 |
ScAAV2 |
NCT00979238 |
|
|
FLT180a |
Free line Therapuetics |
I |
2017-2021 |
AAVS3 |
NCT03369444 |
|
|
SB –FIX |
Sangam |
VII |
2016-2021 |
AAV2/6 |
NCT02695160 |
Hemophilia gene therapy has come a long way in the last ten years. Stable expression of FVIII or FIX has been achieved using wild type or modified FVIII or FIX in an adeno-associated viral vector. Phase III clinical trials have yielded positive outcomes, with patients obtaining trough concentrations of the relevant factor that are adequate to ameliorate the bleeding phenotype. Gene therapy, however, might make it more difficult to monitor hemophiliac patients in a lab setting. The observed disparities between assays following the infusion of conventional or modified factor concentrates, as detailed in this article, are likely to extend to the factor or factors resulting from gene therapy as well. Following gene therapy, coagulation factors (both FIX and FVIII) may be secreted with varying structures and features that may be identified differently by various laboratory assays, hence impacting therapeutic monitoring measures [34]. Variety of cutting-edge techniques, including gene therapy, cell therapy, and regenerative medicine, and tissue engineering, make up advanced therapeutics. They are intended for illnesses or disorders for whom there aren’t a cure at the moment and whose management calls for optimization. International medical organizations state that pharmaceuticals for human use that are based on genes, tissues, or cells and that provide novel approaches to the treatment of certain diseases are the products utilized in the framework of advanced therapies (Figure 1) [35,36].
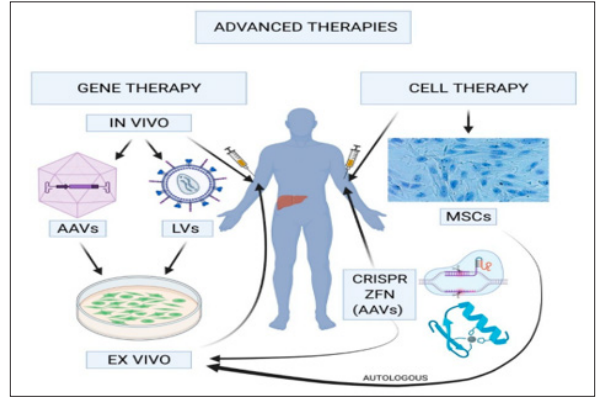
Figure 1: In Vivo Gene Therapy
Novel therapeutic techniques include tissue factor pathway inhibitor (TFPI) with monoclonal antibodies, inhibition of anticoagulation proteins like antithrombin with an RNA interference molecule, and specialized antibodies that imitate the coagulation activity of FVIII (Figure). Furthermore, gene therapy phase 3 clinical trials for hemophilia A and B have started [14,21]. It is used subcutaneously, and regimens of every four weeks, every other week, and every week have all been explored [37].
Hemophilia A and Factor VIII inhibitors in Pediatric Participants: A Study of Emicizumab Administered Subcutaneously) trials included patients with inhibitors who were 12 years of age or older and younger than 12 years old, respectively [38].
Recombinant factor replacement therapy is an effective treatment for hemophilia A and B, significantly reducing morbidity and death.3. Nevertheless, despite factor replacement, such treatment is very expensive and is still plagued by clinical problems, such as bleeding, especially into the joints. To maintain minimal therapeutic levels, recombinant factor concentrates must be administered frequently—two to three times a week—which is a difficult and very intrusive procedure. While highly developed nations have seen some progress in outcomes, most patients worldwide do not have access to the best possible care.1-4 These factors led to efforts to create cutting-edge hemophilia treatment plans utilizing gene therapy, which holds promise for long-term care and possibly even the disease’s cure [37].
The stability profile of FX has significantly improved thanks to newly modified synthetic formulations of FVIII and FIX that are pegylated or fused to long-half-lived proteins like albumin or Fcg. However, the stability profile of FVIII has not improved as much because von Willebr and factor plays a dominant role in determining its half-life. With an improved half-life of $ 38 hours, 5 BIVV001, a new FVIII fusion protein made up of two XTEN linkers and a single-chain recombinant FVIII Fc fused to the FVIII-binding D9D3 domains of von Willebr and factor, offers hope for longer protection in hemophilia. A patient taking medication once a week [39].
Extended half-life medicines allow for a reduction in injection frequency in hemophilia B patients to once weekly or even once monthly, all the while maintaining greater trough levels (FIX. 5%). The use of non-clotting factor products to provide hemostasis in individuals with a bleeding diathesis is another significant advancement. For example, in hemophilia A patients with or without inhibitors, a bi specific antibody (emicizumab) with one arm binding to FIXa and the other to FX enhances the conversion of FX into its active form, leading to a restoration of hemostasis to a degree equivalent to an FVIII level of ~15% of normal [40].
Increasing gene transfer mediated by AAV Considering that AAV delivers the therapeutic transgene as an episome, it is primarily a non-integrating vector. There might just be a minimal integration. Nonetheless, a recent study using a mouse model revealed that the humanized hepatocyte genome integrates at a rate of up to 3% [41]. The current course of treatment involves lifetime intravenous factor IX injections every two to three days to prevent these bleeding episodes. This treatment is invasive, uncomfortable, and non-curative, yet it is good at stopping episodes of spontaneous bleeding. Furthermore, the majority of hemophiliac patients cannot afford the $250,000 annual cost of prophylactic clotting factor concentrate administration, which shortens the life expectancy of individuals with a severe bleeding phenotype. Longer half-lives of novel clotting formulations are a significant advancement, but they still need to be administered intravenously for the rest of one’s life at a considerable expense [42].
Genetic treatment for hemophilia Since hemophilia is a monogenic condition, even minimal expression can result in a phenotypic reversion from severe to moderate, making it an excellent target for the adoption of gene therapy procedures [29,43]. Hemophilia is a chronic illness for which gene therapy is warranted due to the high cost and requirement for frequent factor infusions, which carry potentially deadly dangers. Numerous approaches to hemophilia gene therapy have been put forth. These tactics draw from both ex vivo and in vivo methods. AAV (adeno- associated viral vector) and retroviral are two examples of non- viral or viral vectors used in in vivo delivery studies that have shown extremely positive preclinical evidence and safe early- phase clinical trials [44]. However, obstacles to the safety and efficacy of these strategies—such as the possibility of insertional mutagenesis, undesired immune responses, and possible side effects linked to vector-mediated cytotoxicity—remain in the way of their potential therapeutic success [45]. Therapeutic transgene delivery ex vivo offers a safer approach by preventing viral vectors from spreading throughout the body. In a well-tolerated and safe therapeutic experiment, individuals with severe hemophilia A had autologous skin fibroblasts genetically engineered with the FVIII transgene inserted into their larger tomentum. The survivability of the transplanted cells as well as the FVIII expression levels were believed to be a key barrier to this method, despite the fact that the elevation of FVIII levels was only temporary and small. Hematopoietic stem cell (HSC) utilization and a different method for delivering the therapeutic coagulation factor is offered by autologous endothelial progenitor cells [46,47]. Platelet-based gene therapy, which aims to deliver clotting factors to vascular injury sites by platelets, has been proposed as a new strategy to hemophilia gene therapy recently, and intra articular gene therapy targeting protein expression in affected hemophilic joints [45,48]. Based on the observation that infusion of recombinant human activated factor VII (FVIIa) was beneficial in inducing hemostasis in severe hemophilia, a novel concept for the treatment of hemophilia has emerged recently: continuous expression of activated FVII from a donor gene [49,50]. As a potential transgene, factor VIIa is less likely to elicit an adverse immune response than factor VIII since it regulates hemostasis in hemophilia patients regardless of their usage of F8 inhibitors and all patients should be fully tolerant to it. Future therapy is suggested to use gene therapy as the delivery technique and FVIIa as the transgene [51].
The low expression profile of FVIII and the restricted packaging capacity of AAV vectors (4680 nucleotides) have made it difficult to employ these vectors for hemophilia a gene therapy. The Nathwani group, however, created an AAV-based gene-transfer method that overcomes the limitations of size and ineffective FVIII expression. Removing the FVIII B-domain, this is not necessary for cofactor action allowed the FVIII expression cassette to be smaller. Rearranging the human FVIII wild-type cDNA resulted in a ten-fold increase in human FVIII expression, based on the codon use of highly expressed human genes [2].
Table 2: Characteristics of Approved Factor VIII and IX Products with Extended Plasma Half-Life
|
Proteinname and approval year |
Brand name and manufacturer |
Modification/technology |
Plasma half-life (h) |
Time longer half-life |
|
Efmoroctocog alfa, rFVIII (2014) |
Elocta/Eloctate, Sobi |
Fc-fusion |
19 |
1.5-1.7 |
|
Eftrenonacog al, rFIX (2014) |
Alprolix, Sobi |
Fc-fusion |
82 |
4.3 |
|
Rurioctocog alfa pegol, rFVIII (2015) |
Adynovi/Adynovate, Baxalta/Takeda |
PEGylated (2 × 20 kDa) |
14.3 |
1.3-1.5 |
|
Albutrepenanocog alfa, rFIX (2016) |
Idelvion, CSL Behring |
Albumin-fusion |
101 |
5.3 |
|
Nanocog beta pegol, rFIX (2017) |
Refixia, Novo Nordisk |
GlycoPEGylated (40 kDa) |
93 |
5.8 |
|
Damoctocog alfa pegol, rFVIII (2018) |
Jivi, Bayer |
PEGylated (60 kDa) |
19 |
1.6 |
|
Turoctocog alfa pegol, rFVIII (2018) |
Esperoct, Novo Nordisk |
GlycoPEGylated (40 kDa) |
18.4 |
1.6 |
Fc, fragment crystallizable; PEG, polyethylene glycol; rFVIII, recombinant factor VIII; rFIX, recombinant factor IX [52].
The single-stranded DNA genome of the parvovirus known as Adeno-associated virus (AAV) is approximately 4.7 kb in size. AAV is a widespread natural infection, however it is not linked to any known pathogenic infections in people since it is a dependovirus, meaning it cannot replicate in the absence of a helper virus like an adenovirus. The DNA that codes for the viral protein is removed from recombinant AAVs. The viral genome only contains the inverted terminal repeats (ITRs) needed for packaging, hence AAV vectors can hold a promoter and gene of interest with a packaging capacity of roughly 5 kb [35,53]. Early research on AAV gene therapy for hemophilia B concentrated on skeletal muscle delivery in both people and animal models (mice and dogs) [54,55]. This early research used AAV2 vectors, but more recent clinical efforts for gene transfer to skeletal muscle have used AAV serotype 1 (AAV1) vectors because of their greater transduction capacity in myocytes [56].
Hemophilia B gene therapy has progressed to many recent clinical studies, making it the more effective of the two disorders. This is mostly because F.IX is more straightforward than F. VIII. The 461 amino acid F9 single domain protein is encoded by a 1.4 kb coding sequence. Because of its tiny size, it may be conveniently enclosed in a recombinant adeno-associated virus, a gene therapy vector that has shown promise in treating a range of genetic illnesses [6]. Furthermore, skeletal muscle may successfully perform the posttranslational modification of F. IX, making it possible to conduct early research in a target tissue that is less dangerous than a vital organ like the liver, which is the natural site of F.IX synthesis [14].
Apart from supportive care, transfusions of whole blood or fresh plasma were the only treatments available for hemophilia prior to the 1940s [57]. But none of these can produce enough FIX or FVIII, which is why hemophiliacs will always experience severe bleeding and chronic morbidity. Blood transfusions can only aid patients temporarily and cannot actually extend their lives. The goal of gene therapy is to “edit a defective gene sequence in situ to achieve complete reversion of a disease phenotype for the lifetime of the patient,” which may sound a little lofty [58]. Gene therapy approaches typically concentrate on gene addition rather than gene replacement, despite recent advancements in site-specific rectification of faulty gene sequences. In particular, long-term expression and “expression at levels high enough to ameliorate or cure the clinical phenotype of the disease” are the two main objectives of gene therapy for hereditary diseases [59]. According to recent studies, “adult stem cells, autologous fibroblasts, platelets, and hematopoietic stem cells; by means of non-viral vectors; or through the repair of mutations by chimeric oligonucleotides” can all be treated with gene therapy using lentiviral and adeno-associated vectors [60].
For HA, valoctocogene roxaparvovec (valrox) demonstrated a persistent rise in FVIII activity and clinically significant improvements in bleeding rates [58]. For a number of reasons, hemophilia is regarded as the perfect illness for gene therapy. For example, the therapies available now are costly, inconvenient, and episodic. Preclinical research can be conducted on small and large animal models of hemophilia A and B. More crucially, treatment for the condition can be quantified using well-defined coagulation assays, eliminating a problem that plagues gene therapy efforts for other disorders. Because hemophilia is a monogenic illness that does not require high expression levels for coagulants to take effect, it is also treatable with the more sophisticated cell treatments [61].
Understanding the target cells better is necessary to further the science of gene therapy, such as the hepatocyte’s physiology as a biofactory for FVIII. Understanding the variables affecting a protein’s transcription, translation, post-translation, and release will be crucial for enhancing gene treatments’ effectiveness, safety, and, most importantly, their durability. Future gene replacement treatments need to overcome the obstacles of increasing the therapy’s effectiveness in children and extending the longevity of transgene expression. Because the transgene does not multiply within the cell, transgene expression becomes diluted and eventually disappears with the use of current gene replacement therapies; this is especially true for youngsters. Therapeutics is now working on a CRISPR/Cas9-based in vivo genome-editing program for gene editing. In order to prevent the loss of AAV vector due to hepatocyte growth, this technique uses non-homologous end-joining to enable permanent chromosomal integration of a modified human B-domain-deleted FVIII at the albumin locus in liver cells. When it comes to treating young kids with hemophilia who are not yet able to receive traditional gene therapy, this kind of approach could be revolutionary.
Improved knowledge of liver-targeted gene therapies for additional reasons and liver illnesses, including hepatitis, alcoholic liver disease, and nonalcoholic fatty liver disease, will result from advances in the molecular understanding of transgene insertion into hepatocytes. The ultimate goal of gene therapy is to produce a safe, stable, and long-lasting therapy to replace or supplement missing or defective proteins for a variety of conditions while also reducing the burden on society and patients. Recent advancements in this field are showing promise for the near future.
Since no names, photos, or videos pertaining to specific participants were included in this study, neither ethical approval nor agreement to participate is relevant. Since this university in Ethiopia is very newly established, the researcher just needs a formal communication for all proposed titles from the University management to begin work, unless there was no an ethical board presents on campus.
Consent for publication is not applicable as this study did not include names, images, or videos relating to individual participants.
There is no data collected but we had used different resources studied in the previous for this review.
There is competing interests for the review studied. It might not be appropriate to talk about the conflict of interest that all we writers are agree with one another.
This study did not require funding from anybody or organization.
The review was involved by all authors from title selection to final review writing with cooperation.