Author(s): <p>Vikas Jha*, Tisha Jain, Navdeep Kaur, Mafiz Shaikh, Agraj Bhargava, Mehul Nair, Shruti Narvekar, Divya Dhopeshwarkar, Virag<br /> Gada and Kavita Nalawade</p>
ABSTRACT
HIV can be transmitted from a mother to her child through various means such as blood transfusions, sharing intravenous needles, sexual contact, pregnancy, and breastfeeding. The progression of HIV infection, commonly known as AIDS, involves several stages: viral transmission, acute seroconversion, acute retroviral syndrome, recovery and seroconversion, silent chronic infection, and symptomatic HIV infection. The spread of HIV is considered a pandemic, and since its discovery, an estimated 39 million individuals have lost their lives due to HIV infection, while more than 35 million people are currently living with the disease. Improved medical advancements have led to longer survival for people with HIV, which has contributed to the increased prevalence of HIV/AIDS. Recent studies have focused on understanding the factors that contribute to the varying levels of pathogenicity of the infection, with the aim of finding potential treatments for long-lasting effects. These studies have primarily concentrated on comprehending the defining characteristic of HIV infection, namely the establishment and progression of viral infection through the utilization of its nine major proteins and the complex signaling pathways involved.
The human immunodeficiency virus (HIV), primarily targeting the human immune system, was first detected and isolated in 1983 using lymph nodes from a patient with lymphadenopathy. The infection leads to acquired immunodeficiency syndrome (AIDS) [1]. Characterized by various clinical symptoms, malignancies, opportunistic infections, and declining CD4+ cells and immune functionality. Unfortunately, AIDS is a rapidly spreading disease with a high mortality rate and lacks a cure, only slowing down disease progression. HIV exists in two forms, HIV-1 and HIV-2, both belonging to the Lentivirus genus and Retroviridae family [2]. These viruses are classified based on different antigens and genetic characteristics. HIV-1 is more widespread, with a shorter incubation period and higher transmission rate, while HIV-2 is less infectious and severe, progressing more slowly with milder symptoms and a lower death rate [3]. In this discussion, the focus will be on HIV-1. Inside the viral particle’s central region, the HIV genome consists of two identical single-stranded RNA molecules formed through various steps, including RNA degradation, integration of double-stranded HIV DNA into the human genome, and previral DNA creation [4]. The identification of HIV ignited three decades of productive research into AIDS progression. Early studies, conducted before HIV isolation, provided insights into its impact on CD4+ T cells, systemic immune activation, and immune dysregulation [5, 6]. They also revealed polyclonal B cell activation and inadequate antibody responses to new and previously encountered antigens. Over the past ten years of the AIDS epidemic, significant progress has been made in understanding HIV infection’s pathophysiology, leading to a comprehensive understanding of its clinical course. HIV infection typically progresses through stages over a decade. The primary infection affects 50-70% of individuals, resembling a non-specific clinical syndrome similar to mononucleosis. This is followed by the clinical latency period, lasting a median of ten years, and finally, the clinically apparent AIDS stage characterized by constitutional symptoms and increased susceptibility to neoplasms and opportunistic infections. During the primary infection, HIV presence in lymphoid organs is significant as reservoirs and sites for viral replication. Even during the third clinically latent phase,
HIV persists and progresses without clinical manifestations [7]. Although various anti-HIV antibodies and cytotoxic T lymphocytes (CTLs) targeting different HIV proteins are detected during the primary infection, the immune system eventually fails to eliminate the virus effectively. Human Immunodeficiency Virus type 1 (HIV1) is classified into four evolutionary groups: M (major), N (new), O (outlier), and P [8]. Cross-species transmission, such as from humans to chimpanzees, resulted in Simian Immunodeficiency Virus (SIV) in chimpanzees. HIV-1 group M, responsible for the current pandemic, comprises genetically distinct subtypes (A-D, F-H, J-K, and K), circulating recombinant forms (CRFs), and unique recombinant forms (URFs). The global distribution of HIV-1 subtypes and CRFs is influenced by regional spread driven by socioeconomic and behavioral factors within specific risk groups and ongoing transmission from neighboring regions. Subtype prevalence varies, with Subtype B more common in the Americas, Western Europe, and Australia, Subtype A more prevalent in Eastern Europe and Central Asia, and Subtype C accounting for 46% of worldwide HIV-1 infections, primarily in South Africa and Southeast Asia. Regions with multiple subtypes co-circulate CRFs and URFs, like Central Africa [9].
HIV/AIDS remains a significant global public health issue, with approximately 38 million individuals living with the disease worldwide and 1.2 million in the United States [10]. Unfortunately, less than 50% of HIV-positive individuals receive regular healthcare, and viral load reduction success is limited. Over 200,000 individuals in the United States are unaware of their HIV-positive status. Initiatives like the Ending the HIV Epidemic in the U.S. (EHE) project aim to reduce new HIV infections by 90% by 2030, with increased HIV testing in areas with higher diagnosis rates, especially among risky behaviors. However, the COVID-19 pandemic’s impact on healthcare services, including HIV testing, has been significant as people avoided visits due to COVID-19 risks, leading to decreased HIV cases reported in 2020 compared to 2019, especially in priority populations like Black or African American gay men, Hispanic or Latino gay men, bisexual men, other men who have sex with men (MSM), and transgender individuals in CDC-funded areas.
Despite extensive research, the exact molecular pathways determining whether an infected cell becomes productively infected or enters a latent state remain uncertain. While modern antiretroviral therapy (ART) effectively suppresses HIV and restores immunity, individuals with a history of extensive treatment may struggle to achieve viral suppression due to HIV-1 multi-drug resistance and/or medication intolerance [11, 12]. Such individuals are at considerable risk of clinical progression, morbidity, and mortality, emphasizing the need for innovative ART options to manage their condition and improve outcomes [13]. Through this review, our aim is to gain a deeper understanding of the intricate mechanisms underlying this virus, which has persisted for nearly four decades, and contribute to a more accurate HIV prognosis.
The gp120 protein is of utmost importance in the interaction between HIV-1 and host T cells. Within the viral envelope, 14 spike-like structures comprising three gp120 proteins linked to a gp41 protein facilitate attachment of the virus to host cells [14]. The gp41 protein plays a key role in enabling the spikes’ connection to the viral membrane, facilitating the virus’s binding to host cells [15]. The gp120 component of the spike complex specifically binds to the CD4 receptor on the host cell membrane.
When gp120 interacts with a chemokine receptor on the host cell, it undergoes structural changes that expose the bridging sheet and stabilize its conformation, allowing the viral membrane to fuse with the host membrane and facilitating viral entry into the host cell (Figure 1). The diversity of gp120 loops is crucial for HIV’s ability to evade immune detection, as these loops, with their unique three-dimensional structures, protect the conserved regions of the protein prior to interaction with the CD4 receptor. By evading immune recognition, the virus can persist and continue spreading the disease [16]. HIV relies on the cellular machinery of the host for replication, but the host’s immune responses and intracellular antiviral defenses serve as barriers that hinder virus replication [17,18]. The accessory protein Nef, present in HIV-1, HIV-2, and SIV, plays a vital role in the progression of lentiviral infections, contributing to various interconnected processes that contribute to its in vivo function [19]. Nef’s functions include downregulating the expression of MHC-I and CD4 on the surfaces of infected cells, helping these cells evade recognition by cytotoxic T lymphocytes. Additionally, Nef enhances the activation status of newly infected T cells, rendering them more susceptible to viral replication. In target cells, Nef can also prevent HIV superinfection by suppressing the presence of CCR5 and CD4 on the cell surface [20].
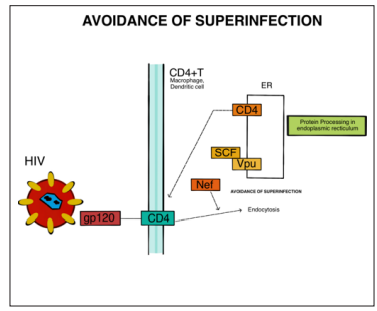
Figure 1: The gp120 Protein of the HIV Viral Envelope Binds to the CD4 Receptor of T Cells, Macrophages, and Dendritic Cells. This Leads to The Activation of Nef, An Accessory Protein of HIV Which is Essential for the Pathogenesis of Lentiviral Infections. Nef decreases Cell-Surface CD4 Levels and MHC-I Expression, to Avoid Being Detected by Cytotoxic T Lymphocytes
The maturation of HIV and its interactions with host proteins heavily rely on protein processing within the endoplasmic reticulum (ER). The endoplasmic reticulum-associated protein degradation (ERAD) quality control mechanism, found in the ER, plays a crucial role in identifying misfolded proteins and directing them towards degradation through the ubiquitin-proteasome pathway [21]. HIV exploits this system to induce the degradation of the plasma membrane protein CD4, which is essential for producing infectious virions during the maturation process [22]. The viral protein Vpu plays dual roles in HIV-1 infection. Firstly, it promotes the ubiquitin-proteasome pathway within the ER, leading to the degradation of newly synthesized CD4 protein. Secondly, Vpu counteracts the host restriction factor tetherin, which inhibits the release of virions from infected cells. By suppressing CD4, Vpu reduces immune responses directed towards infected cells, prevents superinfection of target cells, and enhances viral
replication fitness. Vpu interacts with the transmembrane domains of CD4, facilitating the interaction between TrCP1 and TrCP2 of the SKP1-Cullin1-F-Box (SCF) E3 ubiquitin ligase with CD4 [23]. UFDL1 (ubiquitin fusion degradation 1-like), NPL4 (nuclear protein localization protein), and VCP (valosin-containing protein) are known to interact with CD4. The Vpu protein, along with the SCF-TrCP complex, triggers the activation of the ERAD pathway, leading to the proteasomal degradation of CD4 by removing it from the endoplasmic reticulum (ER). This process is illustrated in (Figure 2) [24, 25].
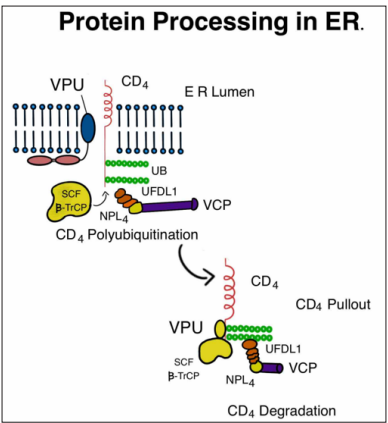
Figure 2: When HIV Binds to the CD4 Receptor, Vpu Binds to the CD4 Receptor, Allowing SCF β-TrCP to Bind to the CD4 Receptor. This Causes the Poly-Ubiquitination of the CD4 Complex and Transport to the ERAD (ER-Associated Degradation) Pathway.
The presence of the primary receptor for HIV, CD4, along with its coreceptors CCR5 and CXCR4, is indispensable for the entry of the virus into host cells. Once HIV gains entry into the target cell, it can activate various signaling pathways to facilitate its replication and entry [26]. Phosphatidylinositol-3,4,5-triphosphates are produced from phosphatidylinositol-4,5-bisphosphate through the action of phosphatidylinositol 3-kinases (PI3Ks). The activation of PI3K is triggered by Gi, a guanine nucleotide-binding protein, as illustrated in (Figure 3) [27]. Phosphorylation, induced by growth factors and hormones, plays a critical role in coordinating multiple cellular processes, including cell growth, cell cycle entry, cell migration, and cell survival. Class I PI3Ks are heterodimeric proteins consisting of a 110 kDa catalytic subunit (p110) and an adaptor/regulatory subunit. These PI3Ks are further categorized into class IA and class IB. Class IB PI3K (p110) functions downstream of G-protein-coupled receptors (GPCRs), while the catalytic subunits (p110, p110, p110) of class IA PI3Ks are activated within receptor tyrosine kinase pathways [28]. Protein kinase B (Akt) is a crucial modulator of PI3K signaling, serving as a serine/threonine kinase. It plays a vital role in regulating various cellular responses, such as cell cycle progression, cell growth, and cell migration. Glycogen synthase kinase 3 (GSK-3) is a downstream target of Akt and another serine/threonine protein kinase that governs physiological reactions to growth stimuli. PI3K is involved in multiple processes, including apoptosis inhibition, down-regulation of MHC (major histocompatibility complex), and the maintenance of HIV-1 latency. Studies have demonstrated that the downstream effectors of PI3K, namely Akt and GSK-3, are indispensable for HIV-1 infectivity [29].
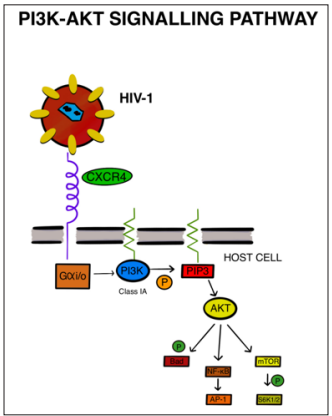
Figure 3: HIV Binding to the CXCR4 Receptor Activates Gαi, Which Is Responsible For The Activation of PI3K. This Catalyzes the Production of Phosphatidylinositol-3,4,5-Triphosphates And Akt Signalling. PI3K Downstream Effectors Are Essential for HIV-1 Infectivity
The mitogen-activated protein kinase (MAPK) cascades are crucial signaling pathways that regulate various cellular processes, including cellular differentiation, apoptosis, and stress responses [30]. Comprising MAP3K, MAP2K, and MAPK, these kinases phosphorylate and activate downstream proteins to transmit signals within the cell. Among these cascades, the Ras/Raf/MAPK (MEK)/ERK signaling pathway stands out as one of the most significant [31]. The extracellular signal-regulated kinases (ERK1 and ERK2) within this pathway, belonging to the MAPK family, play a key role in cell proliferation. Aberrant activation of ERK has been associated with cancer initiation and progression [32]. The pathway is initiated by Ras, an upstream signaling protein, with Raf acting as a MAP3K, MEK as a MAP2K, and ERK as a MAPK. Various stimuli, such as cytokines, viruses, ligands of G-protein-coupled receptors, and oncogenes, can activate the ERK/MAPK signaling pathway through mechanisms like Ca2+ activation, Ras tyrosine kinase receptors activation, PKC-mediated activation, and G protein-coupled receptors activation [33, 34].
Research has indicated the critical role of CD4, the primary HIV receptor, and its coreceptors CCR5 and CXCR4 in viral entry during HIV infection [35]. Stimulation by stromal cell-derived factor 1 (SDF-1) initiates intracellular signaling events, including calcium mobilization and ERK1/2 activation, through the G protein-coupled receptor CXCR4 [36]. G-protein-coupled receptors (GPCRs) interact with Gq and Gi subunits of heterotrimeric G-proteins, leading to the activation of phospholipases C beta (PLC-β), which hydrolyze phosphatidylinositol-4,5-bisphosphate(PIP2) to produce secondary messengers like inositol-1,4,5- trisphosphate (IP3) and diacylglycerol (DAG) [37]. An adaptor protein called Grb2 facilitates the activation of Ras-MAPK by various receptors through its interaction with LAT (Linker for Activation of T cells), ultimately activating the GTPase RAS [38]. The RAS-RAF-MEK-ERK signaling pathway is involved in regulating cell division, proliferation, and survival. Activated RAS interacts with RAF proteins, including C-Raf and B-Raf, initiating the signaling cascade [39, 40]. The Raf family of protein kinases phosphorylates and activates MEK1 and MEK2 MAP kinases, which, in turn, activate ERK kinases [41]. Upon activation, ERK kinases translocate to the nucleus, where they phosphorylate various substrates, including transcription factors, influencing cell survival and other cellular functions [42].
The Calcium signaling pathway plays a pivotal role in both the induction of IL10 by GP120 and the initiation of an immune response [43]. Ca2+ is instrumental in the budding and subsequent release of the viral particle from the host cell. HIV enters CD4+ cells by interacting with chemokine receptors CCR5 and CXCR4. Inside the host cell, the virus engages with proteins like Gaq/11 and Guanine nucleotide-binding protein G(q) subunit alpha, acting as transducers and activators for PLC (phosphatidylinositol phospholipase C), as shown in (Figure 4) [43]. PLC beta, together with receptor tyrosine kinases (RTKs), activates PLC γ, leading to the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into two secondary messengers: inositol trisphosphate (IP3) and diacylglycerol (DAG). Ca2+ molecules bind to the C2 domain of PKCα, β1, β2, and γ subtypes, causing their translocation to the membrane, where DAG binding activates Protein kinase C (PKC), thereby facilitating cell migration. Additionally, Calcium can directly be released into the nucleoplasm through the IP3 receptor. The IP3R protein serves as the primary intracellular Ca2+ storage site, forming a transmembrane calcium ion (Ca2+) channel primarily located on the endoplasmic reticulum (ER) membrane [44].
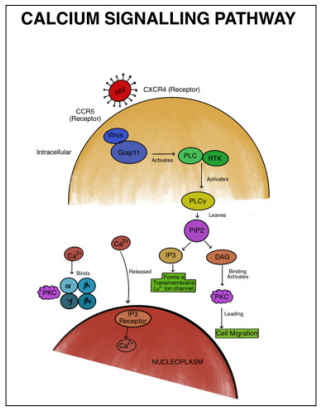
Figure 4: Schematic Summary of Viral Entry in the Host Cells via CCR5 and CXCR4 Leading to a Cascade of Interactions Activating Phosphatidylinositol Phospholipase C With Further Cleavage of PIP2 into IP3 and DA4.
Calmodulin, initially in its inactive form, necessitates activation by intracellular Ca2+ to bind to specific peptide sequences of target proteins. This binding induces structural changes in calmodulin, increasing its activity and enabling its interaction with target proteins. These changes in Ca2+ concentration lead to conformational alterations and activation of calmodulin, which in turn activates downstream target proteins possessing a CaM binding site, including Ca2+/CaM-dependent protein kinases. As a result, calmodulin activity significantly influences the body’s metabolic processes. The Ca2+-sensitive protein phosphatase calcineurin (CaN) also plays a role in this signaling pathway. It activates the transcription factor NFAT, which is critical in the Ca2+ signaling pathway [43]. Abnormalities in the regulation of Ca2+ signaling have been linked to several important human diseases, such as cardiac disease, schizophrenia, bipolar disorder (BD), and Alzheimer’s disease (AD).
Toll-like receptors (TLRs) are a crucial type of pattern recognition receptors (PRRs) involved in the host’s defense against infections. They play a significant role in both the innate and adaptive immune responses by recognizing pathogen-associated molecular patterns (PAMPs). Through coordination with other PRRs, TLRs initiate an immune response aimed at eliminating pathogens and establishing long-term immunological memory [45, 46]. Classified as type I receptors, TLRs are integral transmembrane glycoproteins that belong to a larger superfamily, which includes interleukin-1 receptors (IL-1Rs) [47]. Based on their localization, TLRs can be categorized into two main subfamilies: cell surface TLRs and intracellular TLRs [48]. TLR activation leads to downstream signaling cascades, ultimately resulting in the activation of NF-kB and MAPK, leading to the production of various inflammatory cytokines and chemokines [49]. TLRs also play a critical role in regulating antiviral responses by promoting the production of interferons (IFNs), essential for impeding virus replication and enhancing the immune system’s ability to combat viral infections [50]. However, studies have shown that TLRs can also facilitate HIV-1 infection in innate immune cells through direct mechanisms, such as enhancing HIV-1 transcription or indirectly through cytokine production [51]. TLRs have the capacity to physically interact with HIV-1 components, with TLR8 being identified as the primary sensor for viral single-stranded RNA (ssRNA) in human cells [52, 53].
When TLRs are activated by specific HIV-1 patterns (PAMPs) in infected cells, whether on the cell membrane or in endosomes, they trigger the activation of NF-kB and IRF-3. This results in increased production of immune-modulating cytokines and type 1 interferons (IFNs). Simultaneously, NF-kB activation promotes HIV transcription, while the produced cytokines and IFNs enhance the maturation of dendritic cells (DCs) and influence T cell activation and polarization [54-59]. The specific signaling pathways engaged by TLRs depend on the adaptor molecules involved, with TLR3 and TLR4 activating the TRIF-dependent pathway, while other TLRs engage the MyD88-dependent pathway [60]. The MyD88-dependent pathway primarily leads to the production of inflammatory cytokines, while in specific immune cell types, it may also result in IFN production [61]. MyD88, a critical adaptor molecule in the innate immune response, recruits IRAK kinase family members to form the Myddosome signaling complex. Downstream signaling molecules, including IRAK4, IRAK1, and TRAF6, associate with MyD88, leading to the activation of the protein kinase complex TAK1. TAK1, upon phosphorylation, triggers the IkB kinase-nuclear factor kappalight-chain-enhancer of activated B cells (IKK-NF-kB) pathwayand the mitogen-activated protein kinases (MAPK) pathway, which are crucial for immune and inflammatory signaling, gene regulation, and cellular proliferation (Figure 5) [62-65].
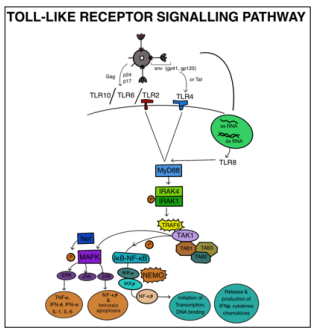
Figure 5: TLR4 Detects Env or Tat While TLR2/6 and TLR10 Detect the Structural Proteins of the HIV Virus, Gag (p17, p24), and Envelope (gp41, gp120), at the Plasma Membrane. Type 1 Interferons (IFNs) and Immune Modifying Cytokines are Expressed More Frequently when a TLR is Activated by Retroviral PAMPs in an Infected cell, Either at the Plasma Membrane or at an Endosomal Membrane. Also, NF-kB Activation Directly Encourages HIV Transcription.
HIV-host cell interactions involve the recruitment of various transcription factors and viral proteins, modulating the transcriptional activity of the HIV-1 long terminal repeat (LTR). Proteins like Tat and Vpu are crucial for full-length mRNA transcript generation in HIV-1. Vpr and Tat exhibit a structural and functional connection, enhancing HIV-1 LTR transcriptional activity and replication [66- 70]. Cellular factors such as Sp1 and NF-kB, along with cytokines, regulate the basal transcription of the HIV-1 LTR. Additionally, cytokines can modulate cellular processes and immune responses in HIV infection [71, 72]. Furthermore, p38 and ERK proteins activated through the MAPK pathway can trigger various cellular responses, including the synthesis of tumor necrosis factor-alpha (TNF-α), which plays a significant role in immune responses and inflammation [73-77]. The presence of specific cytokines in the cellular microenvironment can either suppress or enhance HIV-1 replication and spread in different cell types [78, 79].
The presence of DNA in the cytoplasm can trigger a potent immune response known as the type I interferon response. In the context of HIV infection, specific pattern recognition receptors (PRRs) recognize viral DNA in the cell’s internal compartments and initiate a signaling cascade leading to the production of type I interferons. These interferons play a critical role in antiviral defense by activating immune cells and inducing an antiviral state in neighboring cells. This detection and response to viral DNA through PRRs are essential mechanisms by which the immune system recognizes and combats HIV infection [80]. The immune response triggered by cytoplasmic DNA presence leads to the activation of various antiviral genes, resulting in the synthesis of important antiviral proteins and factors such as interferons (IFNs), APOBEC3, SAMHD1, TRIM5, and BST2. These proteins play crucial roles in inhibiting viral replication and spread. IFNs have potent antiviral properties, limiting viral replication by inducing an antiviral state in infected and neighboring cells. Other factors like APOBEC3, SAMHD1, TRIM5, and BST2 contribute to the host’s defense by targeting viral components and restricting viral replication, collectively contributing to the defense against viral infections, including HIV [81].
During HIV infection, various reverse transcription intermediates, including cDNA, ssDNA, DNA/RNA hybrids, and dsDNA, are produced. These intermediates are essential for the conversion of the viral RNA genome into DNA during viral replication, which occurs within the host cell cytoplasm. The presence of these reverse transcription intermediates can be detected and recognized by the host cell’s innate immune system, triggering antiviral responses and immune activation [82]. Cytoplasmic DNA sensors, such as cyclic GMP-AMP (cGAMP) synthase (cGAS), detect these HIV-generated DNA intermediates. cGAS is an innate immune receptor that binds to cytoplasmic DNA and activates a signaling pathway to initiate an antiviral immune response. Upon binding to DNA, cGAS synthesizes the second messenger molecule cGAMP, which triggers downstream signaling leading to the production of type I interferons and other antiviral factors. The sensing of HIV-generated DNA by cGAS plays a crucial role in the host cell’s defense against viral infections like HIV [83].
cGAS, belonging to the nucleotidyltransferase (NTase) enzyme family, contains structural elements enabling its recognition and binding to cytoplasmic DNA molecules generated during HIV infection. Its enzymatic activity catalyzes the production of the unique isomer of cyclic GMP-AMP (cGAMP), 2’-3’-cGAMP, which acts as a secondary messenger by binding to and activating STING (Stimulator of Interferon Genes) protein. STING, in turn, initiates downstream signaling leading to the activation of innate immune responses against viral infections like HIV [84-86]. Upon activation, TBK1 phosphorylates STING, leading to its dimerization and recruitment of IRF3 (Interferon Regulatory Factor 3), which activates innate immune responses against viral infections. In the context of HIV-1 infection, viral proteins like Vpr and Vpu can either enhance or inhibit the production of type I interferons, impacting the antiviral immune response [87-89]. Host factors, including APOBEC3, SAMHD1, TRIM5, and BST2, act as restriction factors against HIV-1, suppressing viral replication by various mechanisms. SAMHD1, for instance, regulates the levels of deoxynucleotide triphosphates (dNTPs) in the cell, limiting the availability of dNTPs necessary for efficient HIV-1 reverse transcription and preventing productive viral infection (Figure 6) [90-94].
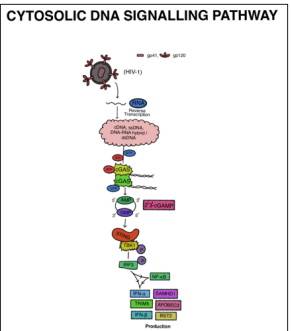
Figure 6: Representation of Steps Involved in the Cytosolic Pathway. Here Reverse Transcription Intermediates are DetectedVia Cgas Further Adhering to ER protein STING Resulting in the Recruitment of TBK1 Followed by TRF3
Various conditions such as inflammatory states, infections, autoimmune diseases, and other disorders can activate immune cells, resulting in the release of inflammatory cytokines like TNF-alpha. Interestingly, the HIV-1 glycoprotein displays molecular mimicry with TNF-alpha, leading to similar effects. Specifically, the HIV envelope glycoprotein GP-120 can stimulate the secretion of TNF-alpha in peripheral blood mononuclear cells. Once released, TNF-alpha binds to the TNFR1 receptor protein present on macrophages and monocytes [95]. TNFR1 is initially maintained in an inactive form by the presence of SODD (Silencer of Death Domain). However, when TNF-alpha binds to TNFR1, it triggers receptor trimerization, which removes SODD and activates TNFR1. The death domain associated with TNFR1 recruits proteins called TRADD and RIP1, which, in turn, recruit TRAF2/5 proteins. This complex activates the IKK (Inhibitor of kappa B kinase) enzyme complex (Figure 7), leading to the phosphorylation of IkappaB. Phosphorylated IkappaB is subsequently degraded, allowing NF-kB to translocate into the cell nucleus. In the nucleus, NF-kB activates the transcription of viral genes like Vpr, Nef, and Tat, leading to the production of functional viral proteins. Additionally, TNF-alpha-induced activation of p38 occurs through dual kinases such as MAP kinase kinases (MKKs), which further contribute to the transcription of Nef and Tat genes [96]. The upregulation of MKKs induces the activation of c-Jun N-terminal kinase (JNK), which, in turn, activates the AP-1 transcription factor. The activation of AP-1 promotes the transcription of genes, including Vpr and Tat, that are crucial for HIV replication. This activation of the HIV LTR (long terminal repeat) facilitates the replication and expression of the virus.

Figure 7: Human Immunodeficiency Virus uses the TNF signalling pathway to generate transcription factor through NFkB translocation, activation of p38, and through C-Jun-N-terminal kinase. Leading to the activation of the HIV LTR activation and thus replication of the virus.
HIV is a viral infection that specifically targets the human immune system, particularly affecting CD4 cells, which are a specific type of white blood cells involved in immune responses [97]. The widespread use of antiretroviral medication (ART) has significantly reduced HIV incidence and transmission by suppressing viral replication. However, it is crucial to understand that ART does not offer a complete cure for HIV, and its use may be associated with unwanted side effects. Furthermore, the virus can persist in certain regions of the body, evading treatment and remaining hidden [98]. The increasing use of antiretroviral medication (ART) has led to the emergence of drug-resistant strains of HIV. While ART can effectively reduce viral replication, it does not completely eliminate the virus. HIV can persist in a dormant state known as the HIV reservoir, even during periods when ART is interrupted. The reservoir contributes to the ongoing transmission of HIV and the progression of AIDS. These latent reservoirs are primarily derived from non-functional CD4+ T cells where viral DNA is integrated, resulting in minimal viral activity within resting infected cells [99]. These reservoirs mainly consist of quiescent memory CD4+ T cells that sustain latent HIV provirus in a transcriptionally inactive state [100].
mTOR, a complex of serine/threonine kinases, plays a crucial role in integrating cellular growth responses to diverse stimuli such as growth factors, hormones, and nutrients in the environment and physiological conditions [101]. mTORC1 regulates various cellular processes, including lipid metabolism, mRNA translation, autophagy, and mitochondrial biosynthesis. On the other hand, mTORC2 controls actin polymerization, cell proliferation, and cell survival [102]. Studies on a primary cell model of Bcl-2 HIV latency and CD4 T cells from patients undergoing highly active antiretroviral therapy (HAART) have shown that mTOR inhibitors such as Torin1 and pp242 can decrease the reactivation of latent HIV when T-cell stimulants are employed [103]. Additional studies have demonstrated that these mTOR inhibitors, when used in CD4 T cells stimulated by CD3/CD28, led to a decrease in global CDK9 phosphorylation. This effect was observed in both uninfected donors and showed a dose-dependent reduction in the activation of the HIV promoter, regardless of the presence or absence of Tat, a viral protein necessary for HIV replication [104].
Recent studies have provided insights into the potential of mTOR inhibitors to hinder acute HIV replication in humanized mice. Specifically, the mTOR inhibitor INK128 has been shown to reduce HIV transcription, as observed in experiments involving U1 cells treated with PMA (phorbol myristate acetate) [105]. Our findings align with previous research, demonstrating that inhibiting both mTORC1 and mTORC2 can effectively suppress the reactivation of latent HIV-1. However, the specific mechanisms responsible for this suppression were not fully explained in the publication. In our study, we investigated latent HIV-1 reactivation in a model using primary CD4 T cells and cells obtained from HIV-infected patients after TCR co-stimulation. We successfully demonstrated that mTOR inhibitors targeting both mTORC1 and mTORC2 can prevent the reactivation of latent HIV-1 in these cell models. The inhibitory effect was also observed in J-Lat and K562 cell lines [106]. In chronically HIV-1 infected primary monocytes and myeloid cell lines, the activation of nuclear factor kappa B (NF-kB) is commonly observed. This activation creates an environment conducive to virus propagation by promoting increased expression of HIV-1 genes within the cells. Elevated intracellular NF-kB levels contribute to the upregulation of viral gene expression, facilitating viral spread [107].
The compound CU-T12-9, acting as a selective agonist of Tolllike receptor 1/2 (TLR1/2), has demonstrated an EC50 of 52.9nM in the HEK-Blue hTLR2 SEAP test. This indicates that CUT12-9 activates TLR1/2 receptors in a dose-dependent manner, with optimal activity observed at approximately 52.9 nM [108]. Through its signaling via NF-kB, CU-T12-9 induces the activation of both innate and adaptive immune responses, leading to the upregulation of downstream effectors such as TNF-α, IL-10, and iNOS. TNF-α is a pro-inflammatory cytokine, IL-10 is an antiinflammatory cytokine, and iNOS is an enzyme involved in nitric oxide production. The activation of these molecules contributes to the immune response and the regulation of inflammatory processes mediated by NF-kB signaling [109]. In contrast, IAXO-102 acts as an antagonist of Toll-like receptor 4 (TLR4) by specifically targeting the co-receptors MD-2 and CD14. This antagonist inhibits the activation of TLR4-dependent proinflammatory proteins by blocking the MAPK and p65 NF-kB signaling pathways. Consequently, the expression of proinflammatory proteins regulated by TLR4 is reduced. IAXO-102 has also exhibited the ability to inhibit the growth of experimental abdominal aortic aneurysms (AAA), suggesting therapeutic potential in treating AAA by modulating TLR4-mediated inflammatory responses [110]. NF-kB, a transcription factor widely present in various cells, plays a crucial role in regulating the expression of numerous genes involved in immune responses and inflammation. Both the canonical and non-canonical NF-kB signaling pathways have been identified as significant factors in the reactivation of latent HIV-1. This indicates that therapeutic strategies targeting these pathways could potentially be effective in disrupting HIV-1 latency and achieving viral suppression. By modulating NF-kB signaling, interventions might effectively control HIV-1 infection and reduce viral reservoirs [111].
The non-canonical NF-kB pathway involves a specific subset of TNF receptors (TNFRs). In the absence of activation, a complex consisting of BIRC2, BIRC3 (also known as cIAP2), TRAF2, and TRAF3 is degraded, leading to the accumulation of Inducing kinase for NF-kB (NIK). Upon receptor activation, BIRC2 and BIRC3 facilitate the transformation and degradation of TRAF3, allowing NIK to accumulate. This accumulation of NIK triggers downstream signaling events, ultimately leading to the activation of the non-canonical NF-kB pathway [112]. Upon NIK activation, IKK alpha is phosphorylated, leading to the proteolysis of p100 and its transformation into p52. The p52 subunit, upon translocation to the nucleus, combines with the RELB transcription factor to form a heterodimer. IAPas (inhibitors of apoptosis protein antagonists) can mimic this process. Furthermore, PKC agonists have been shown to effectively induce independent latent HIV expression, indicating the significance of PKC-NF-kB signaling in preventing HIV latency. This highlights the need for further research and the development of new strategies targeting this pathway [113].
Canonical NF-kB activation involves inducible degradation of IkB through site-specific phosphorylation by a multi-subunit IKK (IKKβ) complex. The IKK complex can be triggered by various stimuli, including cytokines, growth factors, mitogens, microbial components, and stressors [114]. In contrast, the noncanonical NF-kB pathway responds to specific stimuli, such as ligands for certain members of the TNFR superfamily, including LT-βR, BAFFR, CD40, and RANK. This signaling pathway collaborates with the canonical pathway to regulate specific processes within the adaptive immune system [115]. Inhibition of both mTORC1 and mTORC2 has been observed to decrease CDK9 phosphorylation in CD4 T cells activated through TCR co-stimulation, suggesting the involvement of mTOR signaling in CDK9 activity regulation. The precise mechanisms controlling CDK9 phosphorylation and dephosphorylation are not yet fully understood, despite the existence of CDK9-targeting phosphatases (PP1, PP2A, PPM1A, and PPM1G) and CDK9 kinases (CDK2, CDK7, CaMK1D, and CDK9 itself). Further research is needed to understand the intricate regulation of CDK9 and its interplay with mTOR signaling [116].
Suppression of mTORC1/2 can potentially activate additional processes beyond our current understanding due to the diverse effects of mTORC1 and mTORC2. Inhibiting mTORC1 has been shown to promote autophagy. Interestingly, inhibiting mTOR specifically targets Tat, a viral protein crucial for HIV-1 virion production in CD4 T cells. This selective targeting of Tat by mTOR inhibition offers a potential approach to combat HIV replication. Further research is needed to fully understand the mechanisms and implications of mTOR inhibition in the context of HIV infection [117]. Extensive efforts are underway to develop strategies and tools for preventing HIV infection, but achieving this objective still presents challenges. With a growing understanding of mechanisms involved in suppressing active viral replication, maintaining latent infection, or triggering reactivation from latency, there is a current focus on preclinical and clinical studies exploring potential medications that target latent HIV infection. These studies aim to develop novel therapeutic approaches that can effectively control or eradicate the virus during its latent stage.
During the process of synthesizing proteins, intracellular peptides, also referred to as antigens, are produced. These antigens subsequently undergo breakdown by proteasomes. The crucial role of the transporter associated with antigen processing (TAP) is to load these peptides onto major histocompatibility complex class I (MHC-I) molecules and facilitate their transportation into the endoplasmic reticulum. As a defense mechanism to prevent the spread of infection, cells expressing MHC-I molecules complexed with “non-self” peptides undergo lysis. The fully assembled MHC-I molecules are then transported from the Golgi network to the plasma membrane (PM), where they can be recognized by the T cell receptor (TCR) present on CD8+ cytotoxic T lymphocytes (CTLs). The recognition process depends on whether the presented antigens are perceived as “self” or “non-self” [118].
CD8 T-cell activation is reliant on the presentation of antigens through MHC class I molecules. This process is efficiently carried out within the peptide-loading complex situated in the endoplasmic reticulum. Within this complex, a covalent interaction occurs between the thiol oxidoreductase ERp57 and tapasin, which holds significant implications for the peptide-loading process [119]. Recent research progress has provided insights into the interaction between ERp57 and tapasin. ERp57, also known as grp58 and PDIA3, serves as an enzyme involved in facilitating the formation of disulfide bonds in glycoproteins. Elevated levels of ERp57 have been observed in various neurodegenerative diseases linked to endoplasmic reticulum (ER) stress. On the other hand, tapasin plays a critical role as a cofactor in the assembly of MHC class I molecules, ensuring the proper production of peptide-based heavy and light chain heterodimers [120].
Tapasin’s crucial role as a cofactor in MHC class I antigen presentation involves its significant contribution to the synthesis of MHC class I heterodimers. These heterodimers consist of heavy and light chains that are responsible for binding and presentingpeptides. Tapasin facilitates the interaction between the peptidecontaining heavy chain and the light chain, ensuring the proper assembly of MHC class I molecules. This interaction is vital for the formation of functional MHC class I complexes, allowing effective antigen presentation to immune cells [121]. The absence of ERp57 significantly impairs tapasin’s ability to recruit MHC class I molecules into the peptide-loading complex. The interaction between ERp57 and tapasin plays a crucial structural role within this complex, influencing both the recruitment of molecules and the duration of their interactions. While ERp57 is known to function as a thiol oxidoreductase, its precise role within the peptide-loading complex remains a subject of ongoing research, requiring further investigation to fully comprehend its mechanisms and contributions [122].
The HIV Nef protein’s activation has significant implications for HIV replication and the progression to AIDS. Nef serves multiple roles, including immune evasion and interference with antigen presentation. Among its key effects is its impact on the major histocompatibility complex class I (MHC-I) molecules, which play a critical role in presenting viral antigens on the surface of infected cells. Nef disrupts the normal trafficking of MHC-I, resulting in limited antigen presentation at the cell surface. As a consequence, cytotoxic T lymphocytes (CTLs) encounter difficulties in recognizing and eliminating virally infected cells. To develop effective medications for managing HIV infection, it is essential to comprehend how Nef regulates antigen presentation and explore strategies to enhance the capability of anti-HIV CTLs to target the virus [118].
In the early stages of the secretory pathway, Nef interacts with the cytoplasmic tail of MHC-I. Through the utilization of AP-1, Nef alters the normal trafficking route of MHC-I, redirecting it from reaching the plasma membrane to instead enter the endosomal network. As a result of this diversion, MHC-I is prevented from presenting antigens on the cell surface as it would typically do. For MHC-I to continue the degradation process, it must be transported from the endosomal network to late endosomal compartments, where further degradation occurs in lysosomes. This transportation process is facilitated by the combined action of Nef and β-COP [118].
Two proposed hypotheses elucidate how Nef influences the redistribution of MHC-I away from the cell surface. The first hypothesis, referred to as the signaling model of downregulation, involves the host membrane trafficking regulator protein known as phosphofurin acidic cluster sorting protein 2 (PACS-2). According to this model, PACS-2 guides Nef to the trans-Golgi network (TGN). Once Nef reaches the TGN, it activates specific Src-family kinases (SFKs), leading to the endocytosis of MHC-I from the cell surface in a PI3K-dependent manner [123]. Collaborating with membrane adaptor protein-1 (AP-1) and phosphofurin acidic cluster sorting protein 1 (PACS-1), MHC-I’s cytoplasmic tail contributes to sequestering internalized MHC-I within an intracellular compartment. Structural studies have revealed critical interactions between residues Y320 and D327 in the cytoplasmic tail of MHC-I, AP-1, and Nef, highlighting the significance of the cytoplasmic tail of MHC-I in forming the ternary complex with Nef and AP-1 [124].
The second model, termed the stoichiometric form of Nefdependent MHC-I downregulation, gains relevance as the infection progresses. According to this model, Nef interacts with the cytoplasmic tail of MHC-I to direct the membrane trafficking regulators AP-1 and coat protein 1 (COPI), along with the receptor, to a compartment targeted for degradation [124]. These two modes of MHC-I downregulation, the signaling and stoichiometric models, are not mutually exclusive and can coexist during different stages of infection. Additionally, the preference for one mode over the other may depend on the lifespan of the specific cell type involved. For instance, shorter-lived T-cells may be more influenced by the signaling model, while monocytes with longer lifespans may be more affected by the stoichiometric model. These distinct mechanisms of MHC-I downregulation emphasize the complex interplay between the virus and host cells during HIV infection [125].
The viral infectivity factor (Vif) of human immunodeficiency virus type 1 (HIV-1) is responsible for neutralizing the antiviral activity of APOBEC3G (also known as CEM15 or apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G). By reducing the levels of APOBEC3G in HIV-1-infected T cells, Vif prevents its incorporation into viral particles, thereby evading the host’s innate immune response mediated by APOBEC3G [126]. This reduction of APOBEC3G expression is achieved through inhibiting the translation of APOBEC3G mRNA and promoting its posttranslational degradation via the 26S proteasome. The direct interaction between Vif and APOBEC3G is sufficient to cause its depletion, even in the absence of other HIV-1 proteins. Consequently, there is limited incorporation of APOBEC3G into newly formed HIV-1 virions. The absence of Vif severely impairs HIV-1 and other primate lentiviruses’ ability to replicate in most cell culture and in vivo systems. Vif plays a crucial role in facilitating efficient HIV-1 replication in primary CD4+ T cells, which are the natural targets of the infection [127].
Incorporating APOBEC3G into newly produced virions enables it to exert its antiviral activity in the subsequent target cell. APOBEC3G causes specific changes during the reverse transcription process in these target cells, resulting in the conversion of cytosine (dC) to uracil (dU) in the viral minus (initial cDNA) strand of DNA and the substitution of guanine (G) with adenine (A) in the viral plus (genomic) strand. These modifications introduce mutations in the viral genome, impair viral replication, and reduce the infectivity of the newly formed virions [128]. These modifications lead to the termination of the viral life cycle during or shortly after the reverse transcription process. APOBEC3G’s antiviral actions are not restricted to HIV-1 but extend to other susceptible retroviruses, including other primate lentiviruses and endogenous retroviruses. Through the introduction of genetic mutations in the viral genome, APOBEC3G effectively restricts the replication and spread of these viruses, providing a potent innate immune response against viral infections [129].
The antiviral effects of APOBEC3G also extend to other retroviruses such as the equine infectious anemia virus, simian immunodeficiency virus, and murine leukemia virus (MLV). These retroviruses are susceptible to the inhibitory effects of APOBEC3G, leading to the introduction of genetic mutations in their viral genomes during reverse transcription. However, the viral infectivity factor (Vif) collaborates with other processes to counteract the activity of APOBEC3G. By preventing the encapsulation of APOBEC3G into virions and facilitating itsremoval from infected cells, Vif effectively prevents harmful mutations from arising in the viral DNA during reverse transcription in subsequent target cells. This cooperative regulation of Vif and APOBEC3G helps maintain the integrity of the viral genome and promotes successful viral replication (Figure 8) [129].
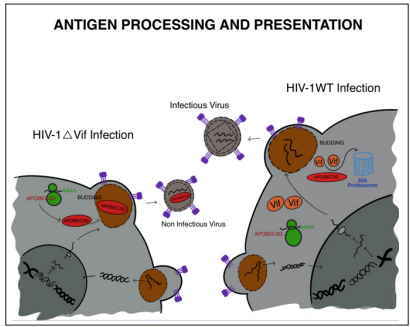
Figure 8: Summary of the Mechanism by which Vif Affects mRNA Translation and Targets the Remaining APOBEC3G Protein for Proteasome Degradation. APOBEC3G is Efficiently Eliminated from the Cell by the Combined Effects of these two Vif Activities, Which Prevents Virions from Encasing APOBEC3G
The cell cycle, which encompasses cell replication and division, is a complex process regulated by numerous proteins. In the case of HIV-1, the virus responsible for AIDS, certain viral proteins, including Vpr, can manipulate the host cell cycle. Similar to HIV1, other viruses like Kaposi sarcoma-associated herpesvirus and Epstein-Barr virus also encode proteins that influence the cell cycle. Understanding the events occurring during the cell cycle is crucial for developing therapeutic strategies against HIV-1 [130].
A significant concern with HIV-1 infection is its ability to establish a latent state within host cells, remaining inactive until it reactivates. Vpr, a viral protein encoded by the HIV-1 genome, plays a crucial role in two aspects of the virus-host cell cycle interaction. Firstly, Vpr disrupts normal cell-cycle control mechanisms, hindering the proliferation of infected cells. Secondly, in conjunction with the matrix protein (MA), Vpr facilitates the entry of HIV-1 into the nucleus of non-dividing cells. This manipulation of the cell cycle by HIV-1 is intricately linked to viral replication and the progression of the virus’s pathogenicity [130]. The cell cycle consists of distinct phases, including S phase for DNA replication and M phase for cell division, separated by G1 and G2 phases. Progression through the cell cycle relies on the activation and inactivation of cyclin-dependent kinases (CDKs) and cyclin regulatory partners. Disruptions in CDK-cyclin complexes can result in cell cycle arrest or other cellular states. HIV-1 manipulates cell cycle progression and alters checkpoint signals to create a favorable environment for viral replication. Vpr and Vif are involved in the G2 arrest of infected cells, halting the cell cycle in the G2 phase. This strategy may be crucial for the virus to modulate its pathogenicity, as viral pathogens often regulate cell cycle proteins. Extensive research on Vpr focuses on its ability to induce G2 arrest, achieved by causing an accumulation of an inactive form of the CDC2 kinase, thus halting the proliferationof infected T cells [131,132].
Vpr engages with various cellular proteins and factors to influence the cell cycle. It stabilizes and enhances the activity of Wee1 kinase, which inhibits Cdc25c phosphatase and keeps Cdk1 inactive. Additionally, Vpr disrupts chromatin’s epigenetic state, leading to premature separation of chromatids and G2 arrest. Furthermore, Vpr targets and activates checkpoint kinases such as ATM/ATR, initiating a phosphorylation cascade contributing to G2 arrest [133, 134]. Viral Protein R (Vpr) is a critical protein encoded by HIV-1 [132]. It plays a vital role in the interaction between HIV-1 and the host cell cycle, disrupting normal cell-cycle control mechanisms and hindering the replication of infected cells. Additionally, Vpr aids HIV-1 in accessing the nucleus of non-dividing cells, demonstrating the intimate link between cell cycle regulation and the proliferation and pathogenicity of HIV-1. The cell cycle comprises various phases, such as G1-S for DNA replication and M phase for DNA division. Progression through the cell cycle relies on the coordinated activation and deactivation of cyclin-dependent kinases (CDKs) in conjunction with their regulatory partners, cyclins [134,135]. Proper coordination of CDK-cyclin complexes is crucial for seamless cell cycle progression. Checkpoints at the G1/S and G2/M transitions act as regulatory mechanisms, pausing the cell cycle in response to abnormal conditions like DNA damage, allowing time for repair. In the context of HIV-1 infection, the virus interferes with cell cycle progression and modifies checkpoint signals to create an environment favoring its replication. This interference impacts various proteins and pathways. For example, HIV-1 activates checkpoint kinases like Chk1 and Chk2, leading to cell cycle arrest at the G1/S phase. During the G2/M phase, HIV-1 proteins, particularly Vif and Vpr, play significant roles in impeding cell cycle progression. Vpr induces G2 arrest by promoting the accumulation of inactive forms of the cyclin-dependent kinase CDC2 and inhibiting the activity of the Cdc25c phosphatase (Figure 9). Additionally, Vpr interacts with chromatin, influencing the epigenetic state, and resulting in premature chromatid separation and G2 arrest [136].
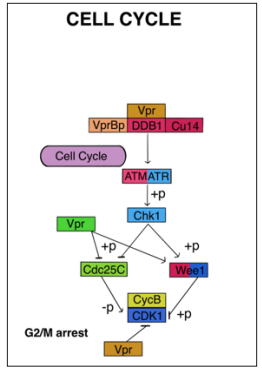
Figure 9: In human Immunodeficiency Virus Type 1 (HIV1)-Infected Cells, a Cell Cycle Arrest in G 2 Increases Viral Expression and May Represent a Strategy for the Virus to Optimize its Expression. Cell Cycle Arrest in G2/M Induced by Vpr has been Reported to Maximize Viral Production Because Of This Stage of the Cell Cycle.
Additionally, HIV-1 proteins, including Vpr and Tat, interact with transcription factors and coactivators to enhance viral transcription. They modulate key proteins involved in the cell cycle, such as p53, Sp1, and NF-kB, to promote viral gene expression. Moreover, Vif plays a role in both viral latency modulation and G2 phase arrest. It stabilizes Wee1 kinase and inhibits Cdc25c phosphatase, resulting in the inhibition of Cdk1 activation and maintaining the cell in a G2 arrest state. The reactivation of latent HIV-1 provirus aligns with T-cell activation and the transition from a quiescent state to active participation in the cell cycle. Vif is believed to be involved in maintaining an active state of p-TEFb, which facilitates viral mRNA production [131]. Consequently, the previously infected cell exhibits increased virus expression. Vif also contributes to G2 phase arrest during HIV-1 infection, as this phase allows for the most efficient transcription of the viral genome. Both Vif and Vpr impact the cell cycle through diverse mechanisms and interactions with cellular proteins. Vif specifically targets APOBEC3 enzymes and PPP2R5 phospho-regulators, while Vpr activates the ATR DNA damage response pathway by promoting the proteasomal degradation of several cellular proteins, including Cullin 4 (CUL4), DNA damage-binding protein 1 (DDB1), and DDB1-CUL4-associated factor 1 (DCAF1) E3-ubiquitin ligase complex. These interactions and modifications orchestrated by HIV-1 proteins ultimately lead to G2/M cell cycle arrest and significantly contribute to viral replication and pathogenesis [137,138].
Apoptosis, a fundamental process of programmed cell death, is critical for cellular development and maintaining homeostasis. It serves as a protective mechanism during immune responses and in the presence of disease-causing pathogens. However, dysregulation of apoptosis has been linked to various medical conditions, including neurodegenerative diseases, ischemic damage, autoimmune disorders, and different types of cancer [139]. In the context of human immunodeficiency virus (HIV) infection, alterations in apoptotic pathways contribute to the progressive depletion of T cells, particularly observed in HIVinfected individuals compared to uninfected individuals. This depletion affects both infected CD4+ and CD8+ T lymphocytes, as well as uninfected bystander T lymphocytes. The apoptosis of uninfected bystander T lymphocytes plays a crucial role in the development of opportunistic infections, chronic inflammation, and the increased risk of malignancies observed in the advanced stage of the disease known as acquired immunodeficiency syndrome (AIDS).
HIV pathogenesis involves the entry of the virus into cells facilitated by the glycoprotein gp120 on the HIV envelope. Gp120 interacts with the CD4 receptor on the cell surface and binds to either CCR5 or CXCR4 chemokine co-receptors, which facilitates viral entry. This process triggers the activation of the CXCR4 receptor on macrophages, leading to increased expression of critical proteins involved in cell death, namely Tumor Necrosis Factor-α (TNF-α) and Tumor Necrosis Factor Receptors (TNFR). These proteins mediate cell death through the extrinsic pathway. TNF-α is a proinflammatory cytokine produced by various immune cells such as macrophages and monocytes in response to pathogen stimulation. It exists in soluble and membrane-bound forms, both of which can activate TNF receptors, initiating downstream signaling pathways that induce cell death [140,141]. The TNF receptors, TNFRI and TNFRII, play roles in activating their respective pathways to induce apoptosis. TNFRI has a death domain in its cytoplasmic region, allowing it to initiate proapoptotic signals within cells. In contrast, TNFRII does not have a death domain and utilizes a different mechanism to induce cell death.
In the context of HIV infection, when the virus attaches to and fuses with macrophages, the presence of TNF-α on the macrophage membrane interacts with the TNFRII receptor on uninfected T cells. This interaction triggers apoptotic death in bystander CD8+ T cells. The binding of TNF-α to TNFRII activates downstream signaling pathways that ultimately result in the apoptosis of the T cells [142]. Moreover, HIV infection is associated with the downregulation of the anti-apoptotic protein BCL-XL, which further promotes cell death. Studies have indicated that TNFRIImediated cell death involves specific factors associated with the receptor, including the TNF receptor-associated factor (TRAF) family of proteins. These TRAF proteins play a crucial role in transmitting downstream signals and modulating cellular responses to TNF-α stimulation, ultimately leading to cell death [140-143].
In the case of TNFRI, the interaction between gp120, soluble TNF-α, and the receptor leads to the activation and trimerization of TNFRI. This trimerization recruits the TNF receptor-associated death domain (TRADD) protein, facilitating protein-protein interactions. TRADD acts as a platform for the recruitment of other proteins in the complex, including Fas-associated death domain (FADD). This complex formation triggers the apoptotic pathway by recruiting caspase 8/10, which subsequently activates executioner caspase 3, leading to cell death.
Another apoptotic pathway involved in HIV pathogenesis is the Fas/Fas Ligand pathway. The Fas receptor (CD95 or Apo-1) belongs to the tumor necrosis factor (TNF) receptor family and contains a death domain (DD) in its cytoplasmic region, which is crucial for promoting apoptosis. This pathway is initiated by the interaction between the Fas receptor and the Fas Ligand, a membrane protein also belonging to the TNF superfamily. The membrane-bound form of Fas Ligand (mFasL) can be cleaved by metalloproteinases, releasing it from the cell surface as a soluble protein (sFasL). mFasL is considered more effective than sFasL in inducing apoptosis. Fas Ligand (FasL) is primarily expressed on the cell surface of immune cells such as lymphocytes, macrophages, and natural killer cells. During HIV infection, Fas Ligand expressed on HIV-specific cytotoxic T lymphocytes (CTLs) interacts with the Fas protein expressed on macrophages, resulting in the recruitment and activation of FADD and TRADD proteins. These proteins then recruit and activate caspase 8, forming a death-inducing signaling complex. Caspase 8 can either directly activate caspase 3, which executes the apoptotic process, or trigger the intrinsic apoptotic pathway.
The intrinsic apoptotic pathway is governed by the BCL-2 family of proteins, consisting of pro-apoptotic and anti-apoptotic members. This pathway is centered around mitochondria. Pro-apoptotic BCL-2 family members, including BAX and BAK, promote the release of mitochondrial factors involved in apoptosis, while antiapoptotic members like BCL-2 and BCL-XL prevent mitochondrial permeabilization and protect against cell death. The balance between these opposing BCL-2 family members ultimately dictates whether the cell undergoes apoptosis [140]. Upon activation, caspase 8 can interact with the pro-apoptotic protein BID, leading to the activation of other proteins such as BAX and BAK. These activated BAX and BAK proteins induce the permeabilization of the outer mitochondrial membrane through their oligomerization, known as mitochondrial outer membrane permeabilization (MOMP), resulting in the release of cytochrome C from mitochondria into the cytosol.
Once released into the cytosol, cytochrome C binds to APAF-1 (Apoptotic Protease Activating Factor-1), forming a heptameric complex called the apoptosome. Within the apoptosome, APAF-1 recruits procaspase 9, leading to its activation through autoactivation. Activated caspase 9 then cleaves and activates caspase 3. Caspase 3, in turn, cleaves its target proteins, ultimately triggering apoptosis of the cell. Additionally, Fas Ligand expressed on macrophages or dendritic cells can interact with cytotoxic T lymphocytes (CTLs) and cause CTL death through the Fas/Fas Ligand pathway.
Apart from host cell factors involved in apoptosis, various viral proteins expressed by HIV, such as Nef, Tat, Vpr, gp120, HIV protease, and Vpu, also play significant roles in influencing cell death mechanisms. Some of these proteins, like Nef, Tat, and Vpu, exhibit anti-apoptotic properties. They interfere with host cell apoptotic signaling pathways, prolonging the survival of infected cells and enhancing viral replication. Conversely, proteins such as Vpr and gp120 have pro-apoptotic effects. They induce cellular stress, activate pro-apoptotic signaling pathways, and contribute to the elimination of infected cells. The interplay between these viral proteins and host cellular factors determines the balance between cell survival and cell death during HIV infection, which can have important implications for viral replication, disease progression, and the host immune response. During the early stages of viral genome replication, HIV expresses the Nef protein at high levels. Nef exerts its influence by suppressing the expression of CD4 and major histocompatibility complex class I (MHC-I) molecules on the surface of infected cells, helping the virus evade the immune response and enhancing its replication.
One crucial function of Nef in HIV infection is to prevent the death of infected cells by inhibiting apoptosis through various pathways. Nef interferes with apoptotic signaling pathways and hinders the activation of pro-apoptotic factors, thereby promoting the survival of infected cells. This enables the virus to persist and continue replicating within these cells. However, it’s noteworthy that Nef can also exert pro-apoptotic effects in uninfected CD4+ T cells. Studies have shown that Nef induces cell death in uninfected CD4+ T cells through the Fas/Fas Ligand pathway. In this pathway, Fas Ligand expressed on infected cells interacts with the Fas receptor on neighboring uninfected CD4+ T cells, leading to their apoptosis. Overall, Nef plays a complex role in modulating apoptosis during HIV infection. It supports the survival of infected cells while also contributing to the death of uninfected CD4+ T cells, thereby influencing the balance between viral replication and the immune response. In uninfected T cells, the infected cells upregulate Fas Ligand (FasL). The Nef protein produced by the infected cells is released in exosomes that can be taken up by neighboring uninfected CD4+ T cells through endocytosis. The uptake of Nef-containing exosomes disrupts proper T cell receptor (TCR) activation, leading to aberrant signaling and ultimately causing the death of the uninfected cells. This process contributes to HIV pathogenesis by depleting uninfected CD4+ T cells, impairing the immune response, and facilitating viral spread. In addition to Nef, the Tat protein, crucial for HIV genome transcription, can also induce apoptosis in uninfected CD4+ T cells. Tat triggers apoptosis through both intrinsic and extrinsic pathways. In the intrinsic pathway, Tat can activate pro-apoptotic factors like Bim, leading to cell death. This pathway involves the disruption of cellular homeostasis and the activation of intracellular signaling cascades promoting apoptosis (Figure 10).
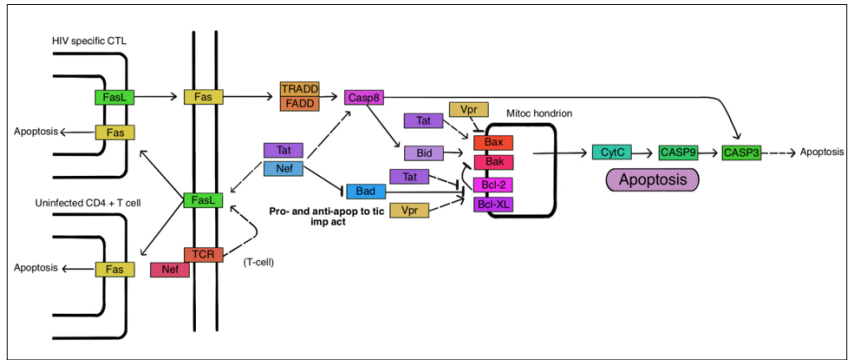
Figure 10: The involvement of HIV Proteins in Intrinsic Apoptotic Pathways: This Protein Is a Target of Tat, Nef, and HIV protease. The Virus Either Alters the Balance of Pro-Apoptotic Bcl-2 Family Proteins to Anti-Apoptotic Counterparts or Vice Versa, Depending on the Pathway Stimulated. HIV Proteins Also Modify Caspase Activation and Regulate Caspase Inhibitors, Which Has an Impact on the p53 Signalling Pathway.
In the extrinsic pathway, Tat can interact with the Fas/Fas Ligand mechanism, similar to Nef. This interaction initiates the extrinsic apoptotic pathway, involving the activation of caspases through the Fas receptor, ultimately leading to cell death. Another protein, Vpr, expressed during the late phase of HIV transcription, possesses the ability to induce and prevent apoptosis in cells. Vpr can induce apoptosis through various mechanisms, including DNA damage response pathways and disruption of the mitochondrial membrane potential. On the other hand, Vpr has also shown anti-apoptotic effects in certain contexts, such as protecting infected cells from apoptosis induced by other viral proteins or cellular factors. These intricate interactions of viral proteins, including Nef, Tat, and Vpr, with cellular signaling pathways contribute to the regulation of apoptosis during HIV infection. The modulation of apoptosisby these proteins is a critical factor in viral pathogenesis, immune evasion, and the overall dynamics of HIV infection.
Ethics approval and consent to participate: Not applicable.
Consent for publication: Not applicable.
Conflict of Interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethical statement: No animals were harmed during this study
Acknowledgement: Not Applicable
Availability of data and material: Not Applicable