Author(s): <p>Kornél Simon</p>
The stress reaction (acute, repetitive or chronic stress patterns) is a non-specific response of an organism to a pathological state. The metabolic characteristics of these stress patterns are well documented. Recognizing the interrelation between a stress-induced pathological state and the relevant pattern of the resultant stress reaction can provide a more accurate assessment of actual cellular metabolism than an approach relying on blood glucose values. Stress activation is obligatorily associated with negative energetic balance in affected cellular metabolism, which is reversed by elimination of the stressor. Prolongation of the stress reaction is a marker indicating the ineffective elimination of the causal stressor. Chronic stress induces progressive self-generating cellular metabolic dysfunction, resulting in the development of both diabetic and ischaemic disorders. Chronic stress conditions are characterized by reductions in inducible metabolic capacity, acute stress tolerance, and functional performance, accompanied by stress-specific micro- and macroscopic alterations. Chronic stress can be activated by both somatic and psychomental stressors. Diabetes, starvation, obesity, ageing, pregnancy, and any chronic disease state of a vital organ are associated with activation of stress reaction. The increased susceptibility of diabetic patients on gliflozins to the development of postoperative euglycaemic ketoacidosis can be explained by a reduction in chronic stress-associated acute stress tolerance, related mainly to diabetes, and a relative glucose deficiency induced by gliflozin therapy. Chronic hyperglycaemia associated with chronic stress should be considered both the primary cause and the secondary consequence of cellular dysmetabolism. Achievement of near-normoglycaemia is a beneficial effect but is not a causal intervention
It is well documented that there is no strong association between blood glucose values and cellular metabolism; therefore, actual blood glucose values, when referred to as a general metabolic parameter, can be accepted only with major reservations [1]. Consequently, proper information on cellular metabolism is clearly desirable. The reason for this expectation is that metabolism has a central position in the whole organism [1-3]. First, the “teleological aim” of the cardiovascular system, respiratory system and tissue respiration is to provide oxygen and supply nutrients for cellular metabolism. Second, the functional and morphological adaptations of organs are supported by the macroergic phosphate energy produced by metabolic processes. Metabolism is served by the “supplying organs” and serves the “executive organs”.
This central role of metabolism is also supported by the fact that the primary cause of all pathological states is the obligatory presence of metabolic disorder (dysmetabolism) in the affected organs. In contrast, the differences between the various pathological states can be determined by the diverse locations of the diseased organs and the very different nature of the harmful stressor agents that evoke the non-specific adaptive mechanism of the organism, which is referred to as the stress reaction by Selye [4, 5]. This mechanism is obligatorily activated in all situations when any harmful agent affects either the somatic or psychomental area of the organism. Although the stress reaction can be triggered by a large number of different agents, resulting in a wide range of pathological states, the types of resultant stress responses can be classified into only a small number of categories, each of which is characterized by well-defined and specific pathophysiological and metabolic parameters. These stress reactions can be viewed as the common unified metabolic responses in the large number of heterogeneous pathological clinical states (Fig. 1). This concept provides the possibility for recognition of the association between any of the large number of pathological states and the relevant stress reaction pattern with similar metabolic characteristics. This approach is expected to generate more information on general and organ-specific cellular metabolism than approaches based on the blood glucose value alone.
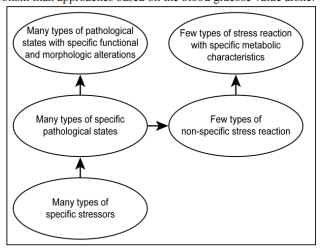
Figure 1: Role of the stress reaction in assessment of the metabolism
The aim of the stress reaction is to eliminate noxious agents or adapt to the stressor effect in order to preserve the integrity and health of the whole organism, i.e., to ensure survival [4, 5]. Stressors can be classified as exogenous (physical, chemical stressors) and endogenous. The latter is exemplified by either an infection of any origin or a psychomental disorder [1, 2, 6]. Exogenous and endogenous stressors often cannot be separated because of their simultaneous existence, e.g., an exogenous stressor can evoke a direct toxic effect and a deleterious emotional reaction at the same time.
The stress reaction can develop abruptly and is referred to as acute stress (e.g., fight
or flight reaction), which can either successfully eliminate the harmful agent or in
contrast, kill the organism. If the stressor effect persists for a prolonged period,the adaptive response also persists, i.e., chronic stress develops, in which not only
membrane receptor-mediated (i.e., functional) but also nuclear-mediated (i.e.,
morphological) alterations governed by both genetic and epigenetic mechanisms
also occur [2, 6, 7]. The phenotypic plasticity phenomenon, i.e., the development of
a disease-specific body composition (e.g., in thyroid, liver, and kidney disorders),
which can clearly be recognized by a visual assessment of the patient (diagnosis
by inspection), is an example in which permanent disease-specific endogenous
stressors are triggers for the development of a specific morphological adaptation
[8, 9].
In chronic stress, inflammatory and immunological reactions inevitably participate
in the adaptive response, which is of obvious relevance in the case of an infection
[2].
Chronic st ress adaptation can also result in the successful elimination of the stressor
or at least in the mitigation of its harmful effect (e.g., increased haemoglobin
synthesis in high-altitude diseases). However, adaptation can sometimes fail when
not only the prolonged stressor effect but also the chronic stress itself contributes
to the long-term damage of the organism (see below).
When the same stressor repeatedly affects the organism, the same functional and morphological adaptations described in chronic stress will occur [2, 6, 7]. This type of stress reaction is referred to as repetitive stress, exemplified by alterations in the cardiovascular system and body musculature that develop following regular physical training. The pathophysiological and metabolic characteristics of stress reaction patterns are summarized below.
The aim of metabolic adaptation in acute stress is to maximize adenosine triphosphate production to achieve the so-called allostatic load, ensuring supernormal metabolic performance in stressed organs, i.e., in the respiratory, circulatory and central nervous systems [2, 6, 7, 10]. The activation of both cytosolic anaerobic glycolysis and mitochondrial aerobic oxidative phosphorylation is an element of this metabolic adaptation [6, 7, 11, 12]. Glucose is the substrate of glycolysis, while acetyl coenzyme A, which originates from both glucose and free fatty acids, is the substrate of metabolic processes resulting in oxidative phosphorylation (Fig. 2). The subsequent metabolic performance is perfectly coordinated: each member of the “metabolic orchestra plays harmonically, guided by the conductor” [6, 7]. Under allostatic loading, both glucose and free fatty acids are metabolized in the mitochondria, with optimal efficiency ensured by a complete oxygen and glucose supply. A continuous glucose supply is supported by glycogenolysis and gluconeogenesis from the liver, and free fatty acids are mobilized from lipid stores. Both hyperglycaemia and increased free fatty acid serum levels are present [6, 13].
A limited oxygen supply is the rate-limiting factor of this maximal metabolic and
functional performance. When this occurs, the substrate consumption in the citric
acid cycle is gradually shifted from free fatty acids to glucose [2]. The reason for
the shift is that the glucose molecule contains more oxygen than free fatty acid;
therefore, its complete breakdown into carbon dioxide and water requires less
exogenous oxygen, i.e., one oxygen atom produces more adenosine triphosphate
molecules when the substrate is glucose than when it is free fatty acid.
It is reasonable to assume that during acute stress, metabolic efficiency can be
induced, i.e., the adenosine triphosphate generation rate per oxygen atom in
oxidative phosphorylation could be augmented. This supposition is in accordance
with the fact that the performance of both the cardiovascular system and tissue
respiration (arteriovenous oxygen difference) also increases during acute stress [2,
10, 14]. The shift from free fatty acid to glucose consumption plays a crucial role
in achieving maximal metabolic performance, while first messengers (adrenaline,
cortisol, glucagon), second messengers (e.g., adenosine monophosphate-activated
protein kinase) and A3 receptors are also important participants in ensuring the
increased cellular uptake of glucose and free fatty acids [1, 2, 10, 13-16].
When a shortage in the glucose supply (due to exhaustion of the glycogenolytic and gluconeogenic capacity of the liver) occurs along with oxygen deficiency during the acute stress reaction, it results in mitochondrial citric acid cycle disorder, i.e., free fatty acid oxidation becomes incomplete, resulting in a tendency to develop ketoacidosis and, later, lactic acidosis. In this state, the residual metabolic activity is mainly limited to cytosolic adenosine triphosphate production of anaerobic glycolytic origin, depending on the amount of cellular glycogen stores. Both adenosine triphosphate generation and the amount of creatine phosphate reserve diminish rapidly: in the absence of an ordered metabolic background, the resultant functional performance cannot be established [2].
In summary, there are two phases in the metabolic adaptation of acute stress. The first is the harmonic, hypermetabolic phase (allostatic load, i.e., eustress), which ensures maximal functional performance. The second is the dysharmonic, disordered metabolic state (allostatic overload, i.e., distress) associated with decreased functional performance. The basic requirement of allostatic load is a perfectly harmonized correlation between the “glucose-related cellular metabolic processes”: cellular glucose uptake, cytosolic anaerobic glycolysis, and mitochondrial aerobic oxidation. In contrast, in the second disharmonic, dysmetabolic phase, malfunction of glucose-dependent mitochondrial aerobic oxidation gradually develops. In allostatic overload, the disharmonic playing of the uncontrolled orchestra results in ineffective metabolic performance, i.e., dysmetabolism [2, 5, 6, 7, 14, 16].
Acute stress is a process characterized by a clearly negative energy balance, i.e.,
the development of an energy-defect state: the creatine phosphate/adenosine
triphosphate ratio continuously decreases in stressed organs. The free oxygen
radical load and calcium ion load of the ultrastructure increase in both the harmonic
phase and even more so in the disharmonic phase of acute stress [2, 6].
During acute stress, along with cellular metabolic events, specific transport
parameters of the blood are present: elevated catecholamine, cortisol, glucagon,
blood glucose and free fatty acid levels [2, 13, 16]. Importantly, a minimal amount
of insulin is also required to achieve maximal metabolic performance in acute stress
[2, 7]. The teleological aim of the supplier serving system (the respiratory and
cardiovascular systems) is to ensure the transport of nutrients and first messengers,
which are indispensable prerequisites in the establishment of increased cellular
metabolic performance.
The metabolic and functional performance of stressed organs can be increased by repeated stress insults, referred to as training [2, 10]. This phenomenon is the result of the training-induced augmentation of the activity of all membrane receptoractivated enzymes involved in both substrate transport and cellular metabolism. The ultrastructural morphological requirements of metabolic hyperfunctioning are also established by activated nuclear receptor-induced de novo protein synthesis, resulting in an alteration of both the number and morphology of mitochondria [2, 6]. In accordance with these functional and ultrastructural adaptations, specific changes occur in cellular substrate consumption: the more advanced the training is, the more dominant the free fatty acid metabolism is in stressed organs, i.e., the shift from glucose to free fatty acid develops [2, 10]. It is logical to assume that the repetitive energy-defect impulses generated by repeated acute stress insults are the primary triggers of this complex metabolic, functional and ultrastructural adaptation [10, 14]. This metabolic state, characterized by repeated generations of energy-defect bursts, is known as “metabolic training” [2, 10]. The shift from glucose to free fatty acid consumption results in greater adenosine triphosphate generation at the molecular level (133 vs. 38 adenosine triphosphate for glucose alone), which, in the case of high and repeated energy demands, can be considered advantageous [2].
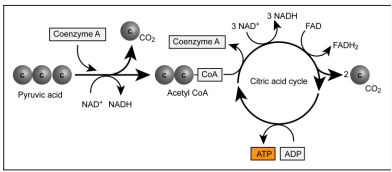
Figure 2: Schematic of the cellular metabolism
Organs with hyperactive metabolism, such as the continuously functioning myocardium and renal and hepatic tissue, are examples of metabolic training in repetitive stress [2, 17]. Interestingly, when the oxygen supply is adequate in the renal cortex, harmonic aerobic mitochondrial metabolism with dominant free fatty acid consumption occurs, while in the hypoxic renal medulla, the main mechanism of adenosine triphosphate production is cytosolic anaerobic glucose metabolism [2].
Another example of repetitive metabolic training is the markedly different metabolic adaptations of sprinters versus marathon runners [2, 13]. In sprinters, repetitive hypoxic insults are generated during repeated acute, extreme exertions performed in a single breath; the energy demand is mainly fulfilled by both anaerobic cytosolic adenosine triphosphate synthesis and the breakdown of cellular creatine phosphate reserves, while significant oxygen and metabolic energy debts are generated. Therefore, in sprinters, training-induced metabolic adaptation is characterized by both an increased replenishment of cytosolic glycogen stores and mitochondrial metabolic training based on the shift from free fatty acids to glucose [2, 13]. For this reason, in sprinters, the main metabolic substrate is glucose.
In contrast, muscular metabolism in marathon runners maintains the energy
balance, i.e., no oxygen debt is generated, and the energy demand is mainly suppliedby mitochondrial oxidative processes based on both glucose and free fatty acid
breakdown simultaneously. The more complete the training is, the more the shift
from glucose to free fatty acid utilization is expressed [2].
The different cellular metabolic patterns of sprinters vs. marathon runners are
necessarily represented in alterations of ultrastructural morphology as well: the
numbers and sizes of mitochondria and the amount of oxygen-reserving myoglobin
in the striated muscles, i.e., the ratio of red vs. white muscle fibres is increased in
marathon runners compared to that in sprinters [2].
It is obvious that the metabolic state of the marathon runner is related to the
allostasis of the steady state, while the sprinter's metabolism is associated with
acute allostatic overload.
As proposed, in the metabolic pattern of repetitive stress adaptation triggered
by repeated metabolic energy-defect insults, the training-induced, augmented,
harmonic, aerobic metabolic performance seems to be associated with dominant
free fatty acid nutrient consumption (substrate switch from glucose to free fatty
acids). This relation might also be interpreted in the reverse direction: the presence
of this type of substrate shift could be viewed as a marker of the metabolic training
state in the background [2, 10].
Although in the repetitive stress-induced hypermetabolic state, the energy supply
is mainly based on free fatty acid consumption, the prerequisite of supernormal
metabolic performance is the complete realization of glucose-related cytosolic
and mitochondrial metabolic processes: “fats burn in the flame of carbohydrates”
[2, 6, 14, 16].
The extent of the increase in stress-related blood glucose and free fatty acid levels is regulated by the amount of exertion [2, 13]. It is also evident that in repetitive stress attacks, the glycogen reserves of stressed organs are repeatedly depleted, and replenishment is ensured by adequately tailored nutrition and rest periods realized in inter-stress intervals. Importantly, the idea of post-exercise hypoglycaemia and hyperinsulinaemia is a well-documented phenomenon in which the replenishing mechanism exists regardless of food intake [2, 13, 18]. During inter-stress rest periods, glycogen and fat stores are replenished, and the cytosolic and mitochondrial ultrastructure required for the higher metabolic performance is regenerated or even further built-up. Both insulin and glucose play crucial roles in this regeneration and renewal process [2, 13]. The potential therapeutic consequence of the latter idea is obvious.
It is also well known that in acute stress, insults with hypermetabolism result in increased free oxygen radical and calcium ion loads in the stressed organs (the brain, myocardium, striated muscles), which inevitably leads to the development of ultrastructural and resultant functional disorders. This phenomenon is more pronounced in allostatic overload than in allostasis. The extent of ultrastructural damage depends on the actual free radical scavenger capacity [1, 2].
It is important to know that the inducible metabolic reserve capacity of trained stress organs can be increased to the maximal level only if the relationship between both the extent and frequency of stress insults and the duration and nutritional characteristics of inter-stress episodes are ideally harmonized. If the parameters of the stress insults and the inter-stress periods are imbalanced, the training results in a paradoxical outcome: the replenishing processes and ultrastructural regeneration cannot successfully be realized, and the metabolic and functional performance cannot improve but instead worsen, i.e., dysharmonic allostatic overload rather than harmonic allostasis could develop [2]. This over-trained state necessarily leads to development of chronic stress activation with all of its deleterious consequences including the diminished tolerance of the acute stress load [2, 19].
The primary and second messenger characteristics of the inter-stress period basically differ from that of the stress reaction. Both are characterised by glucose mobilization, but in the anabolic nutritive state, insulin plays a fundamental role, while in acute stress metabolism of maximal metabolic performance, the main mediators are adrenaline, cortisol, glucagon and adenosine monophosphateactivated protein kinase; however, the role of a limited amount of insulin in achieving allostasis must also be emphasized [2, 6, 13, 16, 18].
If the stressor cannot be successfully eliminated by acute stress adaptation and thus persists, the stress reaction is also prolonged, i.e., chronic stress develops (after 24 hours) [16].
It must be emphasized that while the metabolic changes in acute stress are limited only to the stressed organs, the metabolic changes in chronic stress affect all organs of the organism [2].
The characteristic metabolic changes in chronic stress can be described as follows. Permanently elevated serum levels of cortisol, catecholamines and glucagon inevitably result in the development of insulin resistance [2, 3, 6, 11, 12, 19, 20]. As a consequence, insulin resistance leads gradually to dysfunction in glucose-associated metabolic processes: stress-related elevated serum free fatty acid levels and augmented activity of carnitine acyl transferase lead to markedly increased free fatty acid transport and beta oxidation, which necessarily inhibits glucose-related cytosolic metabolic processes (the Randle cycle) [2, 11]. In chronic stress, substrate consumption shifts significantly to free fatty acids at the expense of glucose [2, 11, 12]. As a consequence, glucose uptake and the replenishing metabolic mechanism referred to as anaplerosis are also damaged [2, 11]. This means that an insufficient amount of oxal-acetic acid is generated from the condensation of pyruvic acid (originating from glucose during cytosolic anaerobic glycolysis) and carbon dioxide. Oxal-acetic acid is a fundamental molecule in the Krebs cycle; its shortage necessarily leads to disordered mitochondrial function and subsequent impairment of adenosine triphosphate synthesis. As a result, acetyl coenzyme A molecules of free fatty acid origin are overproduced and cannot be completely metabolized during terminal oxidation and oxidative phosphorylation; therefore, toxic fatty acids accumulate, resulting in lipotoxicity [1, 2]. The mitochondrial uncoupling phenomenon is also characteristic of this metabolic state: heat production increases at the expense of adenosine triphosphate synthesis, and this occurrence is a marker of inefficient metabolic processes [6, 7, 16, 18]. These chronic stress phenomena inevitably lead to the development of reduced inducible metabolic reserve capacity, ultimately resulting in a decrease in the resting creatine phosphate/adenosine triphosphate ratio. Cellular metabolism is characterized by an increasingly severe metabolic defect, that leads to a reduction in inducible performance and acute stress tolerance and ultimately to damage to resting function [2, 19]. As mitochondrial dysfunction progresses, adenosine triphosphate production is gradually shifted to cytosolic anaerobic glycolysis. Along with the impairment of mitochondrial function, susceptibility to ketoacidosis and lactic acidosis has also been documented, both of which reflect progressive mitochondrial dysfunction. This complex metabolic dysregulation, which is termed the metabolic remodelling state, in chronic stress is strongly similar to the allostatic overload state in acute stress [1, 2]. Moreover, primarily ultrastructural micro- and macroscopic morphological alterations occur in the organs concurrently with metabolic and subsequent functional derangement, followed by the altered phenotype of the whole organism (phenotype plasticity) [2, 6, 7]. These morphological organ and phenotype alterations always show stressor specificity, which is in sharp contrast with the unified, non-specific pattern of metabolic adaptation (Fig. 1).
Increased generation of advanced glycation end-products is also a contributing factor in inducing functional and morphological alterations associated with chronic hyperglycaemia [2].
In summary, in general metabolic disorders caused by chronic stress, the main pathogenetic factor is insulin resistance [1-3]. Alongside insulin resistance, the chronic increase in blood glucose levels and the tendency to manifest diabetes are well documented [1, 2, 19, 20-22]. The glucose toxicity mechanism caused by chronic hyperglycaemia plays a key role in the progressive nature of dysmetabolism: hyperglycaemia further induces insulin resistance, deteriorates beta-cell function, and enhances hepatic glucose output, i.e., hyperglycaemia promotes even more hyperglycaemia. It is evident that in this vicious cycle, hyperglycaemia can be declared both the causal factor (i.e., the “maker”) and the resultant sign (i.e., the “marker”) of cellular metabolic disorder and insulin resistance [23, 1].
It is also logical to suppose that the dysmetabolism of chronic stress most likely affects the organs with the highest metabolic activity, i.e., the vital organs. It is logical to conclude, that acute stress tolerance in organisms with chronic stress should also be depressed.
It can also be supposed that the frequent and marked occurrence of the metabolic
training state, i.e., the presence of chronic stress, disturbs the replenishing processes
during inter-stress periods. As the first and second messenger characteristics of
the acute stress reaction are quite different from those in the inter-stress anabolic
state, the coexistence of these two opposing patterns should be considered a
dysfunctional phenomenon. This means that the lack of energy-defect insults
should be considered the prerequisite of complete replenishment. This proposal
is in accordance with the following statement from Hippocrates: “food is poison
for the sick but is a remedy for the convalescent body” [2].
It is also well documented that in chronic stress, prolonged and marked elevated
levels of serum cardiovascular risk factors inevitably lead to the development of
macro- and microangiopathies, which are necessarily associated with the evolution
of perfusional and metabolic disorders of ischaemic origin [1, 19, 20, 21].
This means that chronic stress can be declared an independent pathogenetic factor
characterized by simultaneous metabolic disorders of both diabetic and ischaemic
origin, which generates a complex, viciously cyclic, self-perpetuating, progressive,
self-damaging harmful process [1, 2, 18] (Fig. 3).
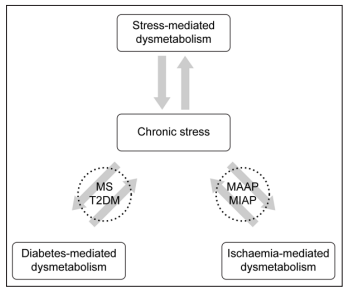
Figure 3: Interrelation between chronic stress, diabetes, and ischaemia
It is important to emphasize that endogenous stressors can also activate the
chronic stress state. It is well documented that psychomental discomfort (e.g.,
civilization stress) can induce the same metabolic, functional, ultrastructural,
micro-/macroscopic cellular and organ alterations as “substantial or material”
stressors [1, 2, 6, 24, 25].
The spectrum of pathological states induced by chronic stress is well documented
and includes cardiovascular manifestations, Alzheimer’s disease and an increased
incidence of malignancies [1, 19, 26, 27].
Importantly, chronic disorders of some vital organs could also be declared primary
stressors. In fact, it is well documented that chronic heart failure, the post-infarction
state, chronic kidney disease, liver disease, and chronic obstructive pulmonary
disease are associated with the long-term development of diabetes [1, 2, 19, 28].
This coincidence could be related to the causal role of chronic stress reactions
induced by chronic pathological organ states [1, 19, 29].
It is also clear that the inducible functional capacity of the organism gradually decreases with age, which can be explained by the age-related decrease in metabolic performance of vital organs as the primary cause and the chronic stress activation evoked by organ dysfunction as the secondary component [30]. These two factors can strengthen each other in a self-perpetuating closed loop.
In patients with diabetes, which is the most common metabolic disorder, the development of metabolic defect states can occur more frequently, but the elimination of stressors can be expected to occur less frequently than in individuals with a nondiabetic status [2, 6]. This led to the proposal that the inducible metabolic reserve capacity in diabetes should be estimated to be subnormal [2, 3, 6, 11].
The repeated multi-organ metabolic-defect insults that occur due to diabetic dysmetabolism can be expected to occur mostly in organs with the highest metabolic activity (the brain, retina, heart, kidneys) [31]. This means that diabetes of any origin is a condition that can be characterized by increased susceptibility to the development of chronic stress [1].
The pathogenesis of type 1 diabetes can be attributed to insulin deficiency. Insulin and the associated glucose-related metabolic processes can be considered the cornerstone mechanism in the achievement of supernormal allostasis. It is clear that insulin deficiency negatively affects the extent of maximal metabolic and functional performance [6, 7].
he pathogenesis of type 2 diabetes is less clear: both insulin resistance and
mitochondrial dysfunction have been proposed as the primary cause [5, 6, 16,
32-34]. Regardless of the primary cause, insulin resistance induces chronic stress
activation, resulting in characteristic alterations of energy-producing cellular
biochemical machinery: glucose-dependent metabolic processes are depressed,
while free fatty acid consumption is augmented. As a consequence of insulin
resistance-induced hypoinsulinaemic effects on cellular glucose uptake, cytosolic
glycolysis and mitochondrial aerobic oxidative phosphorylation, inducible
metabolic performance, i.e., adenosine triphosphate generation, and the resultant
maximal functional capacity, i.e., the acute stress tolerance, is also reduced. (1,
3, 20, 29],
In type 2 diabetes, metabolic syndrome, obesity and old age, the nutrient shift from
glucose to free fatty acid consumption, increased free oxygen radical and calcium
ion generation, and decreased inducible adenosine triphosphate synthesis are well
documented, and all of these processes are in accordance with the reduction in
acute stress-induced maximal functional capacity [2, 6]. For example, in diabetic
patients, the extent of compensatory hyperkinesis in the peri-infarction zone is
markedly diminished [35].
In acute stress, blood glucose elevation is not always present; in moderate stress, hyperglycaemia is occasional; in extreme exertion, however, it always develops [6, 13]. The teleological reason for hyperglycaemia is evident: an adequate glucose supply in stressed organs is a sine qua non requirement to ensure harmonic maximal metabolic performance, i.e., the allostatic load. Ideally, acute stress results in elimination of the stressor effect, i.e., it abolishes the stressor-induced energy-defect state; therefore, the adaptation process can be considered a useful mechanism resulting in the restauration of health [4, 5]. For this reason, the hyperglycaemia of acute stress (i.e., eustress) can be viewed as an absolutely desirable and advantageous phenomenon [2, 5, 13].
It is well known that hyperglycaemia can also occur during acute stress of psychoemotional origin. In this case, it is clear that the reason for hyperglycaemia is to not eliminate the existing metabolic energy-defect state but to prevent its development [2, 30, 36]. It is clear that in this situation, the primary trigger that activates the stress reaction is not the presence of a metabolic defect but that of a subjective psychomental stimulus.
The interpretation of hyperglycaemia in chronic stress requires a rather different evaluation. Chronic stress can be characterized by a dysharmonic, dysregulated metabolism associated with allostatic overload, which is associated with reduced inducible metabolic performance. [2, 6, 19]. In this state, elimination of the harmful stressor has failed, i.e., the permanent generation of metabolic energydefect insults results in the chronic stress reaction. In summary, adaptation of the chronic stress reaction can be considered an ineffective mechanism leading not to recovery but to the development of a self-generated progressive pathological state (i.e., distress) [1, 2, 5, 20, 21].
Hyperglycaemia in chronic stress is a Janus-faced controversial phenomenon. On the one hand, it is a useful tool in acute stress situation that mitigates insulin resistance-induced metabolic disorder; on the other hand, permanent hyperglycaemia induces additional blood glucose elevation by the glucose toxicity mechanism. Therefore, the normalization of hyperglycaemia in a chronic stress state superimposed by an acute stress burden cannot be viewed as a beneficial intervention because counteracting adaptive hyperglycaemia diminishes the actual inducible metabolic performance (2, 4, 5).
An acute blood glucose increase is a marker associated with acute reparative stress; however, a chronic blood glucose elevation is a marker of chronic, selfdevastating stress. As previously suggested, chronic hyperglycaemia (i.e., diabetes) is always associated with the presence of chronic stress. It has been widely proposed that the metabolic dysregulation of diabetes (i.e., diabetic dysmetabolism) is continuously aggravated by cellular overfeeding caused by hyperglycaemia, leading to “cellular glucose poisoning” [37]. It should be emphasized that the direct toxicity of glucose itself cannot be declared the only cause of dysmetabolism because the insulin resistance associated obligatorily with chronic stress also leads to the development of complex metabolic deterioration. This means that hyperglycaemia in diabetes and chronic stress can be viewed as both the secondary consequence and the primary cause of dysmetabolism [1, 6, 38].
It should be emphasized that the basic therapeutic goal of diabetes treatment is the achievement of near normoglycaemia, which counteracts the glucose toxicity mechanism that induces continuous, progressive metabolic deterioration. Notably, this intervention can be considered a causal treatment only for patients with type 1 diabetes, where the basic pathogenetic factor is insulin deficiency [36, 39]. In all other types of diabetes, the principal pathophysiological mechanism is associated either with primary insulin resistance or primary mitochondrial dysfunction. Irrespective of the primary cause, the metabolic consequence results in the activation of chronic stress. It is clear that the evaluation of any antidiabetic intervention should not be limited only to the blood glucose-lowering effect but also to the beneficial or deteriorating influence on the cellular energyproducing biochemical machinery. This means that the wide range of drugs used to achieve normoglycaemia cannot be viewed as equivalent measures in terms of normalization of cellular metabolism [35, 36, 40]. Therefore, new agents of metabolic promotion, such as adenosine monophosphate-activated protein kinase activators, mitokines, and melatonin, which have a variety of subcellular targets in the metabolic machinery, can also be considered potential therapeutic tools beyond currently used interventions [1, 2, 6, 36, 40, 41]. This approach alsoemphasizes that normoglycaemia per se does not obligatorily mean the achievement of eumetabolism. Only the successful elimination of organic or psychomental stressors causing chronic stress activation can be considered causal treatment.
Glucose and free fatty acids are the two main nutritive substrates in cellular metabolism. The main factors influencing the actual cellular substrate consumption can be summarized as follows.
Organ specificity is one of the key determinants of the primary substrate; e.g., in
the myocardium, adipose tissue, and renal cortex, free fatty acids are the preferred
molecule, while in the brain, red blood cells, and renal medulla, glucose is favoured
[2]. The nutritive state always results in a temporary preference for glucose
consumption in all organs, even adipose tissue [2]. Substrate consumption also
changes depending on the functional state of the organ: in the harmonic allostatic
load, free fatty acids are preferred, while in the dysharmonic allostatic overload
glucose is the main source of energy production [6]. Both chronic stress of any
origin (allostatic overload) and the repetitive stress reaction (allostatic load)
can similarly be characterized by predominantly free fatty acid consumption,
although the metabolic efficiency of the two conditions is just the opposite [2].
These latter data clearly show that the actual pattern of fuel consumption (e.g.,
elevated blood glucose value) has no direct association with the energetic balance
of cellular metabolism [1].
Although carbohydrates account for only one-third of the daily caloric intake, glucose molecules still play a central role in energy-providing cellular metabolism [2].
This statement can be supported by the following arguments:
Glucose-related cytosolic and mitochondrial harmonic metabolic processes can establish maximal metabolic performance in the allostatic load [6, 7]. The exceptional relationship between anaplerosis and glucose is the sine qua non prerequisite of maximal efficacy in mitochondrial oxidative phosphorylation [2, 11].
The aerobic oxidation of glucose molecules results in more efficient adenosine triphosphate synthesis than that of any other fuel. Therefore, in any situation characterized by an acute energy demand or a shortage of oxygen, the substrate shift from free fatty acids to glucose always takes place [2, 14]. In the metabolism of malignancies, glucose is also the preferred substrate [2].
In the advanced phase of metabolic remodelling in the chronic stress state, cytosolic anaerobic glycolysis is the last source of adenosine triphosphate generation: glycogen stores are considered the final reserve of active macroergic synthesis [2, 10, 14, 42, 43].
denosine monophosphate-activated protein kinase (referred to as a “metabolic sensor”) is considered the most powerful trigger in the activation of harmonized hypermetabolism, i.e., allostasis [44]. The metabolic sensor activates all glucose-dependent metabolic pathways, such as glucose uptake, glycolysis, and mitochondrial glucose oxidation [6, 10, 14].
Considering all these factors, blood glucose appears to be completely justifiable as the general metabolic parameter. It is also clear, however, that this concept has several limitations in the valid evaluation of the cellular metabolism of the whole organism as well as that of the organs [1].
It has been documented that during starvation and pregnancy and in patients undergoing sodium/glucose cotransporter 2 inhibitor treatment, there are some similarities in terms of metabolic patterns, namely, both the substrate shift from glucose to free fatty acids and the reduced inducible metabolic and functional capacity; furthermore, the propensity for ketoacidosis has been described. Given the coincidence of these metabolic characteristics, chronic stress activation can be suspected [2, 3, 12, 18, 19, 30, 36, 38, 41, 45, 46]. The metabolic correlation between the three above-mentioned states are detailed below.
It is well known that the combination of insulin deficiency, a shortage of glucose supply, and stress activation is the main contributor leading to the development of mitochondrial functional disorders resulting in susceptibility to ketoacidosis and lactic acidosis [6, 13, 38]. The hypoinsulinaemic effect can not only be based on insulin deficiency but also develop as a consequence of insulin resistance.
It is clear why starvation causes glucose deficiency. During pregnancy, the foetal siphon effect can also explain the predisposition to relative glucose deficiency; furthermore, the presence of insulin resistance despite the presence of a hyperinsulinaemic state is an additional documented factor known to induce a relative glucose deficiency in cellular metabolism [35, 38, 46]. As a consequence, a decrease in inducible metabolic and functional performance and a reduction in acute stress tolerance associated with an augmented susceptibility to the generation of metabolic defect can be expected. In accordance with this argument, the development of both asymptomatic acetonaemia and some degree of stress activation have also been well documented during pregnancy [36, 38, 46].
In diabetic patients administered sodium/glucose cotransporter 2 inhibitor treatment, the prevalence of postsurgical euglycaemic diabetic ketoacidosis is more frequent than in those not taking this antidiabetic drug [38, 41, 45, 47-49].
This propensity to develop metabolic defects can be viewed as a sign of reduction in acute stress-induced capacity. It should be emphasized that the proposed chronic stress activation in diabetic patients on gliflozin medication may be related to both the diabetic state and drug treatment. It should be repeatedly stressed that reduced physical performance in diabetic patients subjected to this treatment has also been documented [9, 45, 46].
Further pathophysiological factors hypothesized to participate in the development of an increased susceptibility to postsurgical euglycaemic diabetic ketoacidosis are as follows.
The pathophysiological state induced by gliflozins shows some similarities to pregnancy and starvation. Namely, persistent augmented glycosuria can be viewed as an analogous phenomenon to the foetal siphon effect, as both lead to relative glucose deficiency. Additionally, as a consequence of gliflozin treatment, increased hypoinsulinaemia, hyperglucagonaemia, and insulin resistance have also been documented [38, 41, 45].
The acute stress-induced by surgical intervention is always associated with hyperglycaemia. In the case of glucose deficiency, the increased propensity for the development of ketosis is not surprising. The significance of the preventive replenishment of the cellular glycogen store in the preoperative period should be emphasized [2, 13, 18, 48, 50]. Notably, in this situation, the frequent discontinuation of prior insulin administration has been documented in gliflozintreated diabetic patients with postsurgical euglycaemic ketoacidosis [48]. The increased propensity of gliflozin-treated diabetic patients for postsurgical euglycaemic ketoacidosis strongly supports the concept that euglycaemia is not necessarily associated with eumetabolism [1].
The mechanism underlying the well-documented cardio- and renoprotective
effects of sodium/glucose cotransporter 2 inhibitors has often been discussed
[51]. Inhibition of the glucose toxicity mechanism, resulting in ameliorated
progression of metabolic dysregulation in chronic stress, can be considered the
main contributing factor to the beneficial long-term effects of gliflozins.
In explaining the renoprotective effect of gliflozins, the inhibition of sodium/glucose
reabsorption associated with a very high energy demand could be considered to
effectively promote the metabolic energy balance of proximal tubular cells. This
protective effect is established independent of blood glucose levels.
Recognizing the similarities in metabolic states between pregnancy, starvation,
and diabetic patients treated with sodium/glucose cotransporter 2 inhibitors,
the correlation between these similar metabolic states and chronic stress can
be further viewed as an observation supporting the relevance of stress-based
concepts in the assessment of cellular metabolism compared to those relying on
blood glucose levels.
The main messages of the concept proposed in this paper are as follows: The few types of stress reactions (acute, repetitive and chronic stress patterns) can be viewed as simplified common responses to any pathological clinical state in terms of metabolism. The metabolic characteristics of these patterns of stress reactions are well defined by blood glucose and free acid levels, parameters of first and second messengers, cellular uptake of glucose and free fatty acids, signal transduction mechanisms, glycogen storage, anaerobic energy production occurring in the cytosol, substrate consumption and oxidative energy production of the mitochondria [6, 7, 12, 15, 41].
Identification of the association between any pathological state and the relevant stress reaction pattern functions as a diagnostic tool providing a more accurate assessment of the general metabolic state of the organism and different organs than the blood glucose level alone.
Stress activation obligatorily leads to development of an energy deficit state in metabolism of affected cells. Discontinuation of this metabolic imbalance can be ensured by the elimination of the causal stressor agent, which results in thecomplete functional and morphological restoration of metabolic processes. In this case the non-specific stress activation can be viewed as a successful, healing mechanism (i.e., eustress).
In contrast, when the stress reaction cannot eliminate the harmful stressor, i.e., the trigger is continuously active, the stress activation is also prolonged, which leads to the persistence of the energy deficit metabolic state. This condition is referred to a chronic stress state. Permanent prolongation of chronic stress mandatorily results in evolution of a general complex cellular dysmetabolism, which leads to development of diabetic and ischaemic disorder (Fig. 3). Therefore the continuous chronic stress cannot be viewed as a protective, beneficial mechanism, but on the contrary as a progressively devastating, self-sustaining, self-damaging phenomenon (i.e., distress). This complex pathogenetic mechanism affects mostly organs with the most active metabolism.
Additional findings can be summarized as follows (Table 1):
Chronic stress conditions are inevitably accompanied by a reduction in inducible metabolic capacity, acute stress tolerance, and functional performance. Depending on the nature of the stressors, specific ultrastructural, microscopic and macroscopic alterations can be observed. Table 1. Main conclusions
- Persistence of chronic stress is indicative of the ineffective elimination of the harmful stressor.
- Chronic stress induces permanent progressive, self-generating cellular metabolic dysfunction, resulting in the development of both diabetic and ischaemic disorders.
- Chronic stress is characterized by a reduction in inducible metabolic capacity, acute stress tolerance, and functional performance accompanied by stressor-specific ultrastructural, microscopic, and macroscopic alterations.
- Chronic stress activation can be evoked by not only somatic but also psychomental triggers.
- Diabetes, starvation, obesity, ageing, pregnancy, and any chronic pathological state of any vital organ are necessarily associated with activation of the chronic stress reaction.
- Increased susceptibility of diabetic patients on gliflozins to developing postoperative euglycaemic ketoacidosis can be explained by a reduction in acute stress tolerance associated with chronic stress activation related both to diabetes and the relative glucose deficiency induced by gliflozin therapy.
- Chronic hyperglycaemia associated with chronic stress should be considered both the primary cause and the secondary consequence of cellular dysmetabolism.
-Achievement of near-normoglycaemia in the treatment of chronic stress-induced hyperglycaemia can unequivocally be considered a beneficial intervention but should be viewed a symptomatic treatment. Elimination of the stressor inducing the chronic stress reaction can be considered a causal intervention.
Chronic stress activation can be triggered by not only substantial external and internal triggers but also subjective psychomental disorders. The long-term disease state of a vital organ can also act as a stressor that activates the chronic stress reaction.
Diabetes, starvation, obesity, ageing, and pregnancy alike will inevitably lead to the activation of the chronic stress reaction, resulting in all its undesirable sequelae. The duration of chronic stress is the main determinant of the resultant organ damage, i.e., either reversible or irreversible functional and ultrastructural injuries. The tendency of diabetic patients treated with sodium/glucose cotransporter 2 inhibitors to develop euglycaemic postoperative ketoacidosis can be explained by the reduction in acute stress tolerance associated with the chronic stress, which is related to both diabetes and the relative glucose deficiency associated with gliflozin therapy.
Chronic hyperglycaemia obligatorily bound to chronic stress should be considered both the primary cause of cellular dysmetabolism and the secondary consequence of it, i.e., both a maker and marker of general cellular metabolic disorder.
The normoglycaemia concept in the treatment of chronic stress-induced hyperglycaemia is clearly a beneficial intervention but should be viewed as a symptomatic treatment. Only elimination of the trigger inducing chronic stress can be considered a causal intervention.
Based on the above thesis, novel therapeutic procedures can be proposed in which the duration and nutritional characteristics of the inter-stress period will play prominent roles.