Author(s): Eskedar Kebede Belayneh**, Dagmawi Dereje Wale*, Seid Ibrahim Abdulkadir, Tewodros Kassahun Tarekegn*, Bezawit Girma Gebre, Berhanu Seboka Adugna, Mathewos Tesfaye Yanore, Wedad Abdurahman Muhammed, Tsedenia Ephrem Belay, Dusay Amir Salih, Bezawit Endeshaw Zewde, Beimnet Ayenew Tamene, Belaynew Mekonen Getu and Meron Woldetesay Yilma
Introduction: Insufficient pituitary hormone output is referred to as hypopituitarism. Individuals experiencing lethargy, dizziness, orthostatic hypotension, hypoglycemia, nausea, vomiting, or generalized abdominal pain may be diagnosed with acute adrenocorticotropic hormone (ACTH) insufficiency. This report detailed a unique instance of hypopituitarism in a patient who complained of nausea, vomiting, hypotension, hypoglycemia, and overall abdominal pain and soreness.
Case presentation: A 65-year-old male presented to the outpatient department with complaints of a cough of 1-day duration on August 9/03/21 associated with 4 to 5 episodes of vomiting and pain in the abdomen, in addition to that whole body darkening, fatigue, and appetite loss of 6 months. weight loss intermittent sweating, and eyebrow and axillary hair loss. SOB with mild activity and also bilateral breast pain but no mass.
Discussion: The diagnosis of hypothyroidism and inexplicable fatigue made us believe that there was an adrenal deficiency, which might be treated with cortisol around eight o’clock. Even though the patient with a high stress level the cortisol was immeasurable; so the diagnosis, of adrenal insufficiency, could be made. Other tests including ACTH and Imaging are usually needed to discover the subtype of insufficiency. The follow-up after 1 month showed no signs or symptoms left and a much better condition.
Conclusion: When treating individuals who have fatigue that cannot be attributed to anything else, especially when weight loss and muscle weakness are present, adrenal insufficiency should be taken into consideration. A secondary adrenal insufficiency may exist even in the absence of skin pigmentation, which frequently triggers anxieties and ideas about Addison’s disease. If it is not identified promptly, it may potentially result in an adrenal crisis in situations with high-stress levels. Our patient presented with vague and largely mild symptoms related to early secondary adrenal insufficiency. These included nausea and vomiting, as well as fatigue. Laboratory studies indicated new hyponatremia and urine studies showed inappropriately normal urine sodium and osmolality. His morning serum cortisol was low, with borderline low ACTH, consistent with a secondary cause.
The pituitary gland is divided into anterior and posterior lobes. Anterior pituitary hormones include TSH (thyroid stimulating hormone), LH (luteinizing hormone), FSH (follicle-stimulating hormone), ACTH (adrenocorticotropic hormone), GH (growth hormone), and prolactin. ADH (antidiuretic hormone) is produced by the hypothalamus and travels to the posterior pituitary gland through the pituitary stalk [1]. Hypopituitarism refers to insufficient secretion of the aforementioned pituitary hormones. The diagnosis of hypopituitarism is confirmed via measurement of the levels of those hormones [2].
A severe disorder known as adrenal insufficiency occurs when the adrenal glands are unable to produce sufficient levels of steroid hormones, primarily cortisol, though if the insufficiency is primary, there may also be reduced aldosterone production. All age groups are susceptible, and in Sweden, the frequency is close to one per 100,000 people. In Europe, the principal cause is primarily autoimmunity, while in many underdeveloped nations, the predominant cause is tuberculosis. The cortex of the adrenal glands produces cortisol, aldosterone, and androgens, while the medulla produces adrenalin. The steroid hormone cortisol has a variety of uses, including regulating The immunological response, promoting gluconeogenesis and glycogenolysis, regulating protein metabolism, acting as a stress hormone, and having little impact on water diuresis and sodium retention. whereas aldosterone is a mineralocorticoid hormone that helps in the regulation of blood pressure and the balance of electrolytes. Cortisol secretion is controlled by the pituitary by ACTH hormone, while aldosterone is controlled the renin-angiotensin axis [3-4]. Causes of hypopituitarism include pituitary cell destruction(accountsform orethan95%ofcases), pituitary adenoma [5], hypothalamic tumors (meningiomas and craniopharyngiomas), surgical or radiation therapy of the pituitary adenoma, and infiltrative lesions such as histiocytosis and hereditary hemochromatosis [6]. Patients with ACTH deficiency may present with fatigue, dizziness, orthostatic hypotension, hypoglycemia, nausea, vomiting, or nonspecific abdominal pain [9, 10].
A 65-year-old man arrived at the outpatient department on August 09, 2021 with a one-day cough that was accompanied by four to five episodes of vomiting and stomach pain. He also had entire body darkening, exhaustion, and appetite loss that had persisted for six months. His skin is darkening, he is more tired than usual, and he is losing hair. He has no bulk in his breasts, bilateral breast discomfort, and SOB with light effort. No past medical history of fever, diarrhea, dyspnea, disorientation, or non-recognition. Other symptoms include mood swings, altered sleep patterns, diminished libido, and regular bowel movements. No prior history of problems or obvious disruptions.
No prior history of thyroid issues, diabetes, TB, or alcoholic liver disease. In addition, the patient consumes alcohol three times a week. Other on physical examination, the patient was found to be, slender in build, aware, and alert. Blood pressure of 90/70 mmHg, a low-quantity pulse of 96 bpm, and a breathing rate of 22 breaths per minute. There was no lymphadenopathy, pallor, or swelling in the neck. Hyperpigmentation across the body, primarily on the face, elbows, knuckles, and oral mucosa; no hypopigmented areas have been reported. Examining the abdominal system revealed no guarding, stiffness, or discomfort. fasting blood sugar of 83mg/dL (80-110 mg/dL) SGOT of 60. remaining liver function tests were inside the ordinary range. The erythrocyte sedimentation rate by way of the Westergren method was raised to 40 mm/h (0-7 mm/h Routine examination of urine had no enormous finding. X-ray of the chest and ultrasonography of the abdomen showed no massive abnormality. Due to hypotension, generalized hyperpigmentation, and Hyponatremia in the test results, a differential diagnosis of Addison’s disease was considered. Serum cortisol levels were low at 8 a.m. (serum cortisol = 2.13, ACTH-980). The primary measure of plasma renin within the upright function climbed to 7.76 ng/ml/h (1.90-6 ng/ml/h). When evaluated at rest, the serum aldosterone level was within the lower range of 1.7 (1-16). However, the serum testosterone levels were within the normal range. The levels of plasma ACTH have increased dramatically to about 1250 pg/ml (normally <46 pg/ml). The results of a completed skull X-ray that showed the pituitary fossa were normal. The results of the CT scan show an enlarged adrenal gland and a heterogeneously enhanced left adrenal mass that is ambiguous.
Laboratory Test Value/Unit
| Blood sugar | 40mg/dl |
| AST | 130 U/L |
| ALT | 108 U/L |
| Alb | 3.5 U/L |
| Na | 128 meq/L |
| K | 5.4 meq/L |
| ESR | 38 mm/h |
| Hemoglobin | 13.6 g/dl |
| Platelets | 219 103/L |
| WBC | 6.4 103 /L |
| Triglyceride | 155 mg/dl |
| cholesterol | 150 mg/dl |
| pH | 7.34 |
| Pco2 | 33.7 mmHg |
| Hco3 | 18 mM |
| Pao2 | 104 mmHg |
| O2sat | 99% |
Laboratory Test Value/Unit
| ACTH Level | 980 pmol/L |
| Serum cortisol | 2.13 ng/dl |
Laboratory Test Value/Unit
| Creatine phosphokinase | 3400 mg/dL |
| Lactate dehydrogenase | 1230 mg/dL |
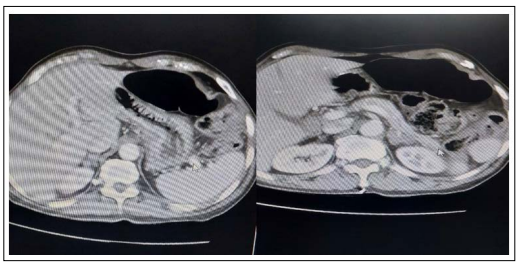
Figure 1: CT Scan
Adrenal insufficiency due to insufficient adrenal corticosteroid, mineralocorticoid, and/or androgen secretion can be caused by diseases of the adrenal gland itself (termed primary adrenal insufficiency), interference with corticotropin (ACTH) secretion by the pituitary gland (termed secondary adrenal insufficiency), or interference with corticotropin-releasing hormone (CRH) secretion by the hypothalamus (termed tertiary adrenal insufficiency). Primary adrenal insufficiency, also known as Addison’s disease, is the most common pathology, but is quite rare, with a prevalence ranging from 39-144 cases per million people [11]. The most common etiology of primary adrenal insufficiency is autoimmune adrenal destruction, which is responsible for up to 70-90% of cases [12]. Approximately 50-65% of patients with autoimmune adrenal disease also have one or more other autoimmune endocrine disorders, including type 1 diabetes mellitus, Graves’ disease, or chronic autoimmune thyroid disease [11,12]. Other causes of primary adrenal insufficiency can include infections (such as tuberculosis, disseminated fungal infection, HIV, syphilis, and African trypanosomiasis), metastatic cancer, adrenal hemorrhage or infarction, and medication-induced damage (including fluconazole, rifampin, and phenytoin, among others) [13-15].
Secondary and tertiary adrenal insufficiency is caused by any disease process affecting the normal secretion of ACTH by the pituitary or CRH by the hypothalamus. Secondary causes, which involve a lack of pituitary ACTH secretion precipitating inadequate adrenal stimulation, include mass effects from pituitary tumors, infectious diseases (such as tuberculosis and histoplasmosis), pituitary infarction, and head trauma [16]. Tertiary causes involve a reduction in CRH secretion, leading to a cascade of inadequate ACTH release and subsequently poor adrenal stimulation. This is most commonly precipitated by the abrupt of high-dose glucocorticoid administration, or following the correction of Cushing’s syndrome. In each of these cases, exposure to supraphysiologic corticosteroid levels for prolonged periods leads the hypothalamus to decrease production of CRH. When the situation is abruptly reversed, the hypothalamus is unable to quickly adjust [17].
Adrenal insufficiency can present in multiple different ways with widely varying severity depending upon the acuity of development, extent of deficiency, and ultimate cause of deficiency (for example central adrenal insufficiency, as in our case, presents somewhat differently from primary adrenal insufficiency).
Symptoms of less acute, more indolent early insufficiency are quite nonspecific. These classically include fatigue (in 85-94% of patients), loss of appetite (53-67%), weight loss (66-76%), nausea, vomiting, and abdominal pain (49-62%), and joint pain (36-40%) [18]. The most common laboratory abnormality is hyponatremia (in 70-80% of patients), followed by hyperkalemia (30-40%), and normocytic anemia (11-15%) of note, in secondary adrenal insufficiency, electrolyte disturbances are less frequent, because the mineralocorticoid axis is typically intact; however, hyponatremia is still commonly seen because of decreased inhibition of ADH caused by a lack of cortisol activity [18]. Evaluation of hyponatremia in these cases will commonly show inappropriately normal urine sodium and osmolality, similar to cases of the syndrome of inappropriate ADH release (SIADH). A classic physical exam finding associated with primary adrenal insufficiency is skin hyperpigmentation (seen in 41-74% of patients), which is caused by excess ACTH leading to increased activation of melanocortin 1 receptors. This is not seen in secondary adrenal insufficiency, in which ACTH is deficient rather than excessive. Because of the non-specific nature of these more indolent presentations, there is commonly a delay in diagnosis. A 2010 cross-sectional study suggested that less than 30% of women and 50% of men ultimately found to have adrenal insufficiency were diagnosed within the first 6 months of symptom onset and 20% of patients had symptoms for 5 years or more before diagnosis [19].
In this setting of non-specific early symptoms and common diagnostic delays, patients are often diagnosed in the setting of an adrenal crisis. An Acute adrenal crisis is generally precipitated in a setting of existing adrenal insufficiency by the development of an additional an acute problem. Classically, a patient with adrenal insufficiency exposed to a new stressor such as acute infection or following a trauma or surgical procedure can develop an acute crisis, as their adrenal function is not able to provide the typical “stress dose” of cortisol needed to meet The physiologic demanded by the event [20]. The hallmark manifestations of an adrenal crisis are dehydration, hypotension, and shock out of proportion to any concurrent known illness. Patients also commonly have other symptoms, including unexplained hypoglycemia, unexplained fever, and acute abdominal pain with or without nausea and vomiting [21]. Without early intervention, the situation can be life-threatening.
Because of the non-specific presentation, making a diagnosis of adrenal insufficiency requires a high index of suspicion. If suspicion is present, the first diagnostic step is to determine whether inappropriately low serum cortisol is present. This can be tested most efficiently with a morning serum cortisol level (ideally measured between 8 and 9 am) [22]. A morning cortisol level over 19 ug/dL rules out adrenal insufficiency and a level under 3 ug/dL effectively rules in adrenal insufficiency.Levels between 4 and 19 require additional testing, typically a 250 mcg corticotropin stimulation test, in which plasma cortisol is measured before administration or 30 to 90 minutes after administration of corticotropin [22]. The test is considered normal if the cortisol level following administration is over 20 ug/dL; the test is abnormal if the cortisol level fails to rise following stimulation. Other tests, such as salivary cortisol have been studied, but are not well-validated and their use on hospitalized patients is less appropriate. Likewise, given the physiologic daytime variation of cortisol levels, single-measurement cortisol levels are only interpretable when drawn in the morning [22].
Once a diagnosis of adrenal insufficiency is confirmed, the next step is the establishment of the level of defect within the hypothalamic-pituitary-adrenal axis (i.e. primary vs secondary vs tertiary insufficiency). Plasma corticotropin (ACTH) level, commonly checked at the same time as a corticotropin stimulation test, is often sufficient. Primary adrenal insufficiency will be characterized by high ACTH levels, whereas secondary and tertiary insufficiency will involve low or inappropriately normal ACTH levels [22].
In the case of mild or early secondary adrenal insufficiency, equivocal results can be seen. In this instance, either the insulin tolerance test or the metyrapone test may be indicated for further investigation. The insulin tolerance test monitors physiologic stimulation of cortisol release in the setting of insulin-induced hypoglycemia [23]. This test is limited by the need for extensive monitoring and its unpleasantness to the patient. The metyrapone test involves the administration of metyrapone, which blocks the last step in cortisol synthesis (from 11-deoxycortisol to cortisol), artificially lowering serum cortisol [22]. Following metyrapone administration, serum 11-deoxycortisol level and serum ACTH are measured - in the case of normal patients the levels will rise significantly, whereas, in a setting of adrenal insufficiency, they will not rise or will rise only marginally. Given the complexity of the administration of these tests, their use is best accomplished with endocrinological consultation. Distinguishing between secondary and tertiary adrenal insufficiency can commonly be accomplished using clinical context. However, if necessary, a corticotropinreleasing hormone (CRH) stimulation test can be performed. Following administration of CRH, patients with tertiary adrenal insufficiency will have a large increase in ACTH levels, whereas those with secondary adrenal insufficiency will have little to no response [24].
Once the level of axis defect is known, a final diagnostic step is a determination of the ultimate cause of the pathology. For primary adrenal insufficiency, workup will likely involve abdominal imaging, measurement of autoantibodies to evaluate an autoimmune cause, consideration of tissue sampling and further infectious workup as indicated by context [25]. Determining the cause of secondary or tertiary adrenal insufficiency will typically involve brain imaging initially, with further workup guided by clinical context.
The treatment of adrenal insufficiency is aimed at replacing the deficient hormones. The backbone of treatment is corticosteroid replacement, with the goal being mimicking of normal diurnal cortisol rhythm. A common regimen involves hydrocortisone, dosed two or three times daily, dosed as low as possible to maintain symptom relief [25]. Patients with primary adrenal insufficiency will also often require mineralocorticoid replacement to prevent sodium and intravascular volume loss as well as hyperkalemia. Hydrocortisone has some mineralocorticoid activity, but patients commonly also receive an agent such as fludrocortisone, typically dosed daily [25]. Likewise, with primary adrenal insufficiency, some patients (especially female patients) have been noted to have decreased mood and quality of life, which is suspected to be related to androgen deficiencies. In these cases, supplementation with daily DHEA has been shown to have modest benefits in mood and quality of life [26].
Finally, a mainstay of long-term treatment of adrenal insufficiency involves patient and family education, with special importance placed upon recognition of the risks and limitations of long-term replacement regimens. Even with adequate long-term regimens, patients will remain at constant risk for adrenal crises should they develop additional stress-related situations, including infections, surgical procedures, and traumas. Patients and their families must understand this risk and how to mitigate it. With minor infections, patients can often independently increase steroid dosing for short periods to mimic the adrenal stress response [25]. In other more severe instances, patients may require physician-guided dose adjustment. In the setting of acute hospitalization, treatment teams will commonly need to provide stress doses of steroids as indicated by the clinical context [25].
When treating individuals who have fatigue that cannot be attributed to anything else, especially when weight loss and muscle weakness are present, adrenal insufficiency should be taken into consideration. A secondary adrenal insufficiency may exist even in the absence of skin pigmentation, which frequently triggers anxieties and ideas about Addison’s disease. If it is not identified promptly, it may potentially result in an adrenal crisis in situations with high-stress levels. Cortisol analysis at 8 a.m. can occasionally be used to identify it, although the Synachten test is more frequently used. Additional testing for adrenal insufficiency was not necessary because the patient experienced an adrenal crisis, was under stress, had low cortisol, and neither of these conditions was present.
Our patient presented with vague and largely mild symptoms related to early secondary adrenal insufficiency. These included nausea and vomiting, as well as fatigue. Laboratory studies indicated new hyponatremia and urine studies showed inappropriately normal urine sodium and osmolality. His morning serum cortisol was low, with borderline low ACTH, consistent with a secondary cause. Given the high clinical suspicion given the context, the adrenal insufficiency was recognized with a minimum diagnostic delay, and the patient was able to be effectively treated with medical supplementation. He will require continued surveillance of symptoms and laboratory studies to guide future treatment adjustments and he will need to be continuously aware of the risks associated with adrenal insufficiency.
None
The authors have not declared a specific grant for this research from any funding agency in the public, commercial, or not-forprofit sectors.
The customers’ written informed consent was obtained, including permission to publish this information and the usage of photos.
None