Author(s): Pier Maria Fornasari
Based on Chinese CDCP report on COVID-19, 14% of patients presented severe disease and 5% critical conditions. The average case-fatality rate was 2.3%, but mortality was as high as 49% in patients with critical illness. Serious life threatening thromboembolic complications have been found in 71•4% of non survivors and micro/macro angiopathic coagulopathy has been found, at autopsy also, with highly increased neutrophil number, fibrinogen, concentrations of D-dimer and FDPs and NETs, ATIII decrease and normal number of platelets. A cytokine storm and interaction between inflammation and coagulation has been advocated as explanation of hypercoagulability.
It has been shown that SARS-CoV-2 infection of alveolar cells is driven by the S-protein by engaging ACE2 and TMPRSS2 cell receptors. whose activation depends on the activity of various host proteases. Full inhibition of SARS-CoV-2 entry was observed when serine proteases inhibitor camostat mesylate was coupled with Cathepsin B/L inhibitor E-64d.
In addition multiple proteases are involved in host immune response against viral invasion and immunopathology related to imbalanced immune activation. In this paper it’s hypothesised that the severity of Covid-19 is induced by recruitment of innate responder neutrophils, which release proteases and NETs inducing endothelial damage and imbalance of the four major proteolytic cascades (coagulation, complement, fibrinolysis and kallikrein) with prevalence of activators over inhibitors and consequent thrombotic complications. Platelets adhesion to damaged endothelium and vWFVIII multimers presence, due to loss of ADAMTS13, contributes to hypercoagulability state. Human plasma or serine protease inhibitors like aprotinin can help to control neutrophil induced “proteolytic storm”.
The goal of this paper is to support the view that, in SARS-CoV-2 infection, proteases have a key role and exceeding imbalanced neutrophil innate “unfriendly fire” response can be identified as the trigger of a “proteolytic storm”, responsible for subsequent well known hypercoagulation and “cytokine storm” and human plasma, in adequate volumes, together with serine proteases inhibitors can be an effective therapeutic strategy.
According to the largest current report from the Chinese Center for Disease Control and Prevention with 72 314 cases, 58 574 patients (81%) were classified as mild, 10 124 (14%) were classified as severe, and 3616 (5%) were considered critical (respiratory failure, septic shock, and/or multiple organ failure) [1]. Among 201 patients in Wuhan, Wu reported that risk factors associated with development of acute respiratory distress syndrome and death included older age, neutrophilia, organ dysfunction, coagulopathy and elevated D-dimer levels [2].
As of February 16, 2021, John Hopkins Covid-19 dashboard has documented a total of 109.211.000 cases with over 2.410.000 deaths worldwide.
The SARS-CoV-2 infection is a protease dependent process as cell entry depends on the binding of the Spike protein’s S1 subunit to ACE2 on the target cell surface and host proteases, furin, as well as TMPRSS2 processes the S protein to facilitate membrane fusion, allowing SARS-CoV-2 to have enhanced proteolytic activation in a wider range of tissues [3].
The strategy used by SARS-CoV-2 for cellular entry is the same in all the tissues presenting dual-positive ACE2+TMPRSS2+ cells, employing its S protein, previously primed by TMPRSS2 protease and cathepsin B/L, binding to ACE2 receptor [4-8]. ACE2, the viral receptor, and one of its entry-associated proteases, TMPRSS2, are expressed (Figure: 1) in nasal goblet cells, in lung goblet, multiciliated and AT2 cells and gut epithelial enterocytes, in pancreatic ductal cells, bladder, testis, prostate, and kidney epithelial cells, cholangiocytes, oligodendrocytes in the brain, inhibitory enteric neurons, heart fibroblasts/pericytes, and fibroblasts and pericytes in multiple other tissues. In line with the kidney’s role in the renin-angiotensin-aldosterone system, dual-positive cells are enriched in the proximal tubular cells and in principal cells of the collecting duct [9].
A recent paper has shown that therapeutic aprotinin concentrations inhibit SARS-CoV-2 replication as entry inhibitors and by compensating for downregulated cellular protease inhibitors during later replication cycles. Aprotinin aerosol, as approved in Russia for the treatment of influenza, may be a particularly promising strategy to suppress virus replication and thus prevent Covid-19 lung injury [10,11].
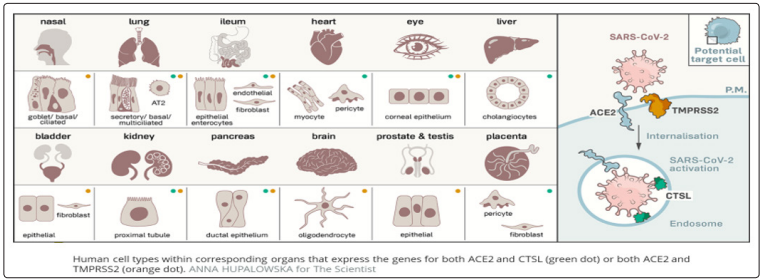
Figure 1: Human Cell Types Potential SARS-CoV-2 Targets Due to Dual Expression of ACE2 Receptor and TMPRSS2
Severe Covid-19: Profound Immunologic ImbalanceInverse correlation between disease severity and lymphopenia has been observed according to Tan L critical patients with lymphocyte percentage < 5% were more likely to become critically ill, with need for intensive care therapy and high mortality rate [12]. Along the same line, imbalance between interferon production and chemokines and an “eicosanoid storm” have been found.
The physiological response to virus infection starts at intracellular replication, through pattern recognition receptors (PRRs) and transcription factors (interferon regulatory factors (IRFs) and nuclear factor kB) activation, inducing cellular antiviral defenses (IFN-I and IFN-III, respectively) and subsequent upregulation of ISGs and leukocytes recruitment by chemokine secretion [12-14].
Imbalance Between Interferon and ChemokinesEarly interferon antiviral activity is depressed, while innate immunity neutrophils are highly recruited. The infected cells delay the IFN-I and -III response by inhibiting innate immune signaling, induce upregulation of chemoattractants for neutrophils and monocytes (HMGB1, CCL2, CCL8 and CXCL family), cytokines storm (IL-2R, IL1RA, IL-6, IL-8, IL-10, and TNF) and increased reactants biomarkers (e.g., procalcitonin, serum ferritin, and C-reactive protein) [15].
The combination of TNF-α and IFN-γ induced inflammatory cell death characterized by pyroptosis, apoptosis, and necroptosis (PANoptosis). Mechanistically, TNF-α and IFN-γ co-treatment activated the JAK/STAT1/IRF1 axis, inducing nitric oxide production and driving caspase8/FADD-mediated PANoptosis. TNF-α and IFN-γ caused a lethal cytokine shock in mice that mirrors the tissue damage and inflammation of COVID-19 and inhibiting PANoptosis protected mice from this pathology and death [16].
Proteomic and metabolomic studies showed activation of complement pathways, acute phase reactants (C-Reactive Protein and Serum Amyloid proteins SAA1 and SAA2), proteins implicated in interleukin IL-6 signalling, Inter-α-Trypsin Inhibitor Heavy Chain 4 (ITIH4), haptoglobin (HP), Leucine-Rich Alpha2-Glycoprotein (LRG1), Monocyte Differentiation Antigen CD14 and the Liposaccharide Binding Protein (LBP), known to induce IL-6 expression. Thus, the proteomic approach surprisingly revealed a very IL-6 centered response.
SARS-Cov-2 infected lung cells also overexpressed complement activation genes, involved in neutrophil degranulation, with deposits of terminal complement components C5b-9, C4d and MASP2 attacking the host ECs and causing transmembrane channel formation on the endothelium and inducing endotheliopathy, involved in ARDS like syndrome with systemic inflammation and lung neutrophilia [17].
An upregulation of fibrinogen, Protein Z-Dependent Protease Inhibitor and SERPINA10 was found, further highlighting the importance of coagulation in SARS-CoV-2 infection.
In severe COVID-19 lung infection, a catastrophic microvascular injury syndrome is caused by activation of complement and coagulation pathways, inducing an associated procoagulant state [18].
Imbalance Between Innate Immunity Cells (Figure 2)Qin described the occurrence of a dysregulated immune response with a marked decrease in T-cell number, higher leukocyte counts and neutrophil-to-lymphocyte ratio (NLR), as well as lower percentages of monocytes, eosinophils, and basophils [19].
SARS-CoV-2 nsp9 and nsp10 directly target NKRF to facilitate IL-8/IL-6 production and thus the response is imbalanced versus activation of the innate immune response [20]. Innate immune response plays an important protective or destructive role, depending on the progression of the disease. The association between human serine proteases trypsin, thrombin, tryptase, and elastase with increased expression of MCP-1 has been shown and the inhibition of these proteases resulted in the inhibition of MCP-1 secretion via inactivation of various protease-activated receptors (PARs) [21]. Furthermore, neutrophils respond to viral infections by activation of neutrophil elastase, Cathepsin G, and proteinase 3, playing roles both intracellularly and extracellularly [22, 23].
However, the presence of a high number of neutrophils at the inflammation site could correspond to imbalanced protease activity. This further leads to various inflammatory disorders, tissue damage, lung dystrophy, ARDS, and potentially death [23].
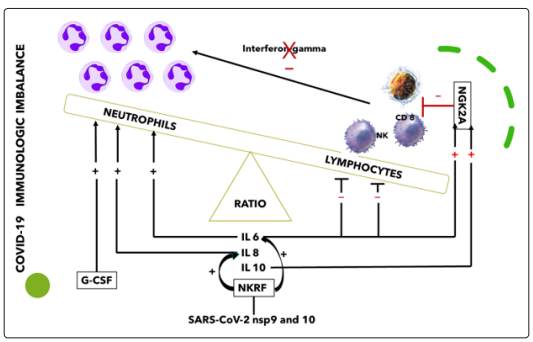
Figure 2: Innate Immunity Cells Imbalance ( Personally Modified by Antonioli L. et al
Several cytokines, such as IL6 and IL-10, were shown to upregulate NKG2A expression and consequent lymphocyte downregulation, while IL-6 and IL-8 impair the functions of NK cells via STAT3- dependent mechanisms [24]. NKG2A, thus, is a key factor in the altered balance between neutrophils and lymphocytes (Figure: 2).
High levels of IL-6, IL-8 and G-CSF enhance neutrophil recruitment and express an inhibitory action on NK cells further reducing interferon (IFN)-γ production [24-26]. Recent studies and autopsy results have confirmedinfection and destruction of lymphocytes in the spleen, lymph nodes, and lymphoid tissues of the gut [27,28]. In these sites lymphocytes were reduced markedly in germinal centers and in situ hybridization detected SARS viral positivity in the residual immune cells in the spleen and in circulation.
Focusing on the areas of the respiratory tract involved and based on the cells that are likely infected, COVID-19 can be divided into three phases that correspond to different clinical stages of the disease.
Stage 1: Asymptomatic state (Initial 1-2 days of infection): inhaled virus SARS-CoV-2 likely binds to epithelial cells in the nasal cavity and starts replicating. The viral burden may be low.
Stage 2: Upper airway and conducting airway response (Next few days): virus propagates and migrates down the respiratory tract along the conducting airways.
Stage 3: Alveolar phase. The disease COVID-19 is clinically manifest. About 19% of the infected patients will progress to stage 3 disease and will develop pulmonary infiltrates. The virus infects alveolar type II cells. SARSCoV-2 propagates within type II cells, large numbers of viral particles are released and the cells undergo apoptosis and die.
The alveolar phase is evolving in 2 different patterns: Moderate with absent or minor endothelial leakage,
Severe with alveolar collapse due to surfactant loss, fluid filling of interstitium, engulfing protein-rich fluid with neutrophils release products like NETs, reduced gas exchange, endothelial lesion, through which SARS-CoV-2 virus can enter into the bloodstream and induce viral sepsis.
In SARS-Cov-2 infected lung cells, deposits of terminal complement components C5b-9, C4d and MASP2 attack the host ECs and cause transmembrane channel formation on the endothelium and induce endotheliopathy, involved in ARDS like syndrome with systemic inflammation and lung neutrophilia [25]. A similar pattern has been shown in purpuric skin lesions due to a pauci-inflammatory thrombogenic vasculopathy, with colocalization of COVID-19 spike glycoproteins and deposition of C5b-9 and C4d in both grossly involved and normally-appearing skin and in lung interalveolar septa [29].
Platelets also play a critical role in lung innate defense response, being lung a primary site for platelet biogenesis: activated platelets engulf virions and secrete antiviral molecules (eg, a-granules) to destroy virions, HMGB1 and express surface P-selectin enabling the initial attachment of neutrophils from the bloodstream [30]. Figure: 3
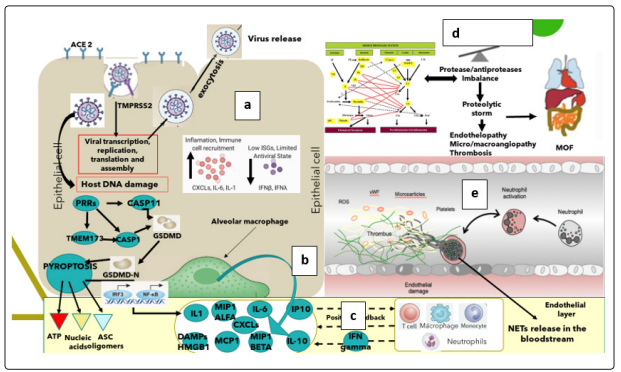
Figure 3: a) Viral infection, b) activation of inflammatory response proteins,c) neutrophil recruitment, d) proteolytic cascades imbalance, e) endotheliopathy and NETS formation
The lung alveoli are thus the last defense line against SARSCov-2 bloodstream dissemination and viral sepsis and thus innate immunology is highly involved in fighting. In severe COVID-19, neutrophils, together with other mononuclear, are the first cells of the immune system to migrate to the infected alveoli, recruited by interferons, IL-1β and IL-6, where they attack SARS-CoV-2, but in addition to direct virus-inflicted pathologies, their exaggerated “unfriendly fire” responses, resulting in a “proteolytic storm” and, inducing alveolar capillary endotheliopathy, contribute to disease severity and subsequent viral dissemination [15,31].
The mechanisms that neutrophils undertake for host defense are phagocytosis, degranulation, cytokine production and neutrophil extracellular traps (NETs) release, known as NETosis.
Neutrophil extracellular traps are DNA structures released including histones and over 30 components of primary and secondary granules, such as elastase, myeloperoxidase, cathepsin G (CG), lactoferrin, pentraxin 3, gelatinase, proteinase 3, LL37 and peptidoglycan-binding proteins.
Three models for NETosis are known to date: suicidal (NETs release and neutrophil lysis), vital NETosis (triggered by TLRs stimuli, platelet glycoprotein Ib, complement activation, after release neutrophils are still able to phagocytose pathogens and have a normal lifespan) and mitochondrial DNA is released instead of nuclear DNA [32].
Platelet HMGB1 protein (passively released extracellularly as a prototypical DAMP from dying cells or stressed or activated cells present in any tissue) is the major endogenous inducer of NETs formation. NET chromatin disrupts epithelial lining, induces platelet aggregation and activates further neutrophils recruitment. NETs, via electrostatic interactions, activate the contact pathway of coagulation and, through tissue factor, the intrinsic pathway [31, 32,33]. NETs form a scaffold for thrombus formation by promoting platelet adhesion and by concentrating coagulation factors involved in clotting. Thrombus-resident neutrophils are strategic for thrombi extension by binding factor XII and supporting its activation through NETosis [34].
The endothelial cells damage (endotheliopathy), induced by SARS-CoV-2 virus and neutrophil elastase, triggers the activation of two independent endothelial pathways (inflammatory and microthrombotic), through release of inflammatory cytokines (interleukin [IL]-1, IL-6, tumor necrosis factor-α, and others) and activation of the platelet and endothelial exocytosis of ULVWF, mediating microthrombogenesis via “activation of microthrombotic pathway”. In parallel endothelial damage inhibits ADAMTS13 biosynthesis, while Neutrophil Elastase proteolytically cleaves and significantly decreases its plasma level [35]. FXIa and α-thrombin remove C-terminal domain of ADAMTS13, blocking its ability to cleave VWF on the endothelial cell surface, and increase the release of VWF antigen by endothelial cells, resulting in persistence of VWF strands and causing an increase in platelet adhesion under flow conditions [36].
This pathological chain of events event has been described also for multisystemic vasculitis in Kawasaki Disease, characterized by platelet stimulation with increase in the shedding of Pselectin, translocating at the surface and externalized with subsequent hyperactivation, and the detection of circulating platelets- neutrophils aggregates. Neutrophils recruited by platelets Pselectin and PSGL-1 (vascular adhesion molecules playing an important role in the inflammatory response by mediating the interaction of leucocytes with stimulated endothelium and platelets bound in the vicinity of vascular injury) contribute, through Toll-like receptor 4, to NETs formation, NETs cause platelet activation and aggregation, thus linking inflammation and thrombosis to support the relevance of this mechanism in the pathogenesis of Covid-19.
Simultaneously, serine proteases released by neutrophils and present in NETs cleave coagulation inhibitors such as tissue factor pathway inhibitor and antithrombin [37].
On the damaged endothelial surface NETs, ULVWF multimers, platelets and activated not inhibited clotting and complement pathways initiate thrombogenesis within the microvasculature, leading to microthrombi enriched also by leukocytes recruited in the P-selectin dependent manner [38]. The microthrombi can become sufficiently large to be released from endothelial cells into the circulation, resulting embolism [39]. This condition can be recalled “TTP-like syndrome” [40].
Blood circulation action and mechanical stress (like forced ventilation) may be sufficient to physically disrupt the fragile structure of NETs in the bloodstream, releasing NET fragments. Mechanically disrupted NETs augment NETosis and NETosis propagates inflammatory response.
TLR inhibitors may reduce inflammation, specifically by preventing NET-induced NETosis
Intravascular NETosis is thus responsible for initiation, dissemination and local accretion of thrombotic events in arteries, veins and in the microvasculature, with end-organ damage in lungs, heart, kidneys and other organs [41].
Through endothelial lesions and NETs, SARS-CoV-2 virus activates its viremic phase and in each interested organ (Figure: 1) follows the same cellular entry strategy and catastrophic microvascular injury syndrome, causing final MOF.
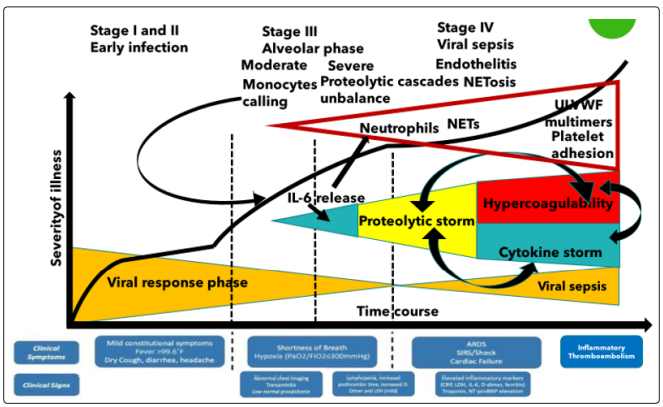
Figure 4: Covid-19 clinical evolution. Neutrophils are actively recruited by cytokines and platelets, released high levels of serine proteases imbalance mainly coagulation/complement/fibrinolytic cascades inducing a severe “proteolytic storm” with hypercoagulability (micro/macroangiopathic endotheliopathy), endothelial lesions and inflammation, NETs formation and bloodstream diffusion through mechanical fragmentation. Through endothelial lesions, SARS-CoV-2 can disseminate to organs presenting dual ACE2 receptor and TMPRSS2
If NETs induce hypercoagulability, the significant increase in neutrophil numbers and the released proteolytic enzymes (mainly elastase) contribute to a consumption of proteases inhibitors, with an umbalance of physiologic conditions and instauration of the “proteolytic storm” [42].
The activators/inhibitors balance in proteolytic cascades is essential for homeostasis and, due to this, in normality the inhibitor plate is largely superior to activator one [43-45] .
The innate serine protease system has four major columns, coagulation, fibrinolysis, kallikrein and complement [46]. These systems are strictly correlated, interconnected and their physiological mantainance is the result of a rigorous balance. Complement directly enhances coagulation and, in addition, inhibits anticoagulant factors, while certain coagulation enzymes activate complement components. The interplay between complement and coagulation is crucial to understand the clinical implications in Covid-19, in which complement-coagulation interactions contribute to the development of life-threatening complications [22].
The contact system, also named as plasma kallikrein-kinin system, consists of three serine proteinases: coagulation factors XII and XI, plasma prekallikrein and high molecular weight kininogen. Once activated by NETs, this system is prothrombotic by activating intrinsic pathway and proinflammatory by producing bioactive peptide bradykinin.
Extrinsic and intrinsic pathway of blood coagulation induce simultaneous activation of the complement and fibrinolysis cascades, with an extensive cross talk mutually fine-tuning their activation status [47].
Main family of the serine protease inhibitors (SERPINs) is formed by SERPINA1 (𝔞1-antitrypsin) protecting lung tissue from neutrophil elastase, SERPINA5 (Protein C inhibitor), SERPINC1 (also known as antithrombin) controls coagulation proteases, SERPIND (Heparin cofactorII), SERPINE1 (plaminogen activator inhibitor 1), SERPING1 (also known as plasma C1 inhibitor) regulates complement, callicrein and contact phase activation and SERPINF2 (also known as 𝔞-2- antiplasmin) inhibits plasmin and regulates fibrinolysis. Complement activation is inhibited also by Decay-accelerating factor (DAF) and Factor H (alternative pathway). Alpha 2 macroglobulin acts as an antiprotease for a variety of proteases like plasmin, kallikrein and thrombin. A delicate balance between serine proteases and their serpin inhibitors is crucial for normal functioning of biological pathways [48].
Proteases/antiproteases balance is present also at the endothelial surface, where thrombomodulin, forming complexes with thrombin, induces protein C activation to suppress blood coagulation, while TNF-alpha and IL-1beta, inducing TF and PAI1, down-regulate the expression of thrombomodulin. Procoagulant TF upregulation with downregulation of the anticoagulant TM/ Protein C system converts the normal anticoagulant endothelium into a prothrombotic endothelium.
Lastly, NETs triggered significant platelet aggregation. A proteases/antiproteases balance is present also at alveolar space, where SERPINA1 strongly and specifically inhibits neutrophil elastase. When the inhibitor concentration is sufficient to block released elastase, no lesion happens nor in alveolar epithelium nor in alveolar endothelial wall and this corresponds with the moderate Covid-19 clinical condition [49].
Otherwise, if SERPINA is overhelmed or is absent/deficient, as in homo/eterozygous patients (about 4% European population), imbalance between elastase and anti-elastase activity, free elastase causes progressive damage of both alveoli and endothelium, inducing endotheliopathy and thrombogenic state previously described [50].
The hypercoagulability, as unbalance of proteases/antiproteases cascades, the decrease of ADAMTS13, the endotheliopathy, the increased platelet activation, the ULVWF multimers and the NETosis together create a severe thromboembolic environment, similar to Thrombotic Thrombocytopenic Purpura/HUS conditions and Acute Promyelocytic Leukemia activation of clotting systems with secondary hyperfibrinolysis [51-53].
Convalescent Plasma (CP) use in the therapy of untreatable infectious diseases has been extensively but anecdotally documented, including spanish Influenza A (H1N1) infections in 1915 to 1917, severe acute respiratory syndrome (SARS) in 2003, pandemic 2009 influenza A (H1N1), avian influenza A (H5N1) and several hemorrhagic fevers such as Ebola. Based on studies, showing convalescent plasma antibodies can limit the virus reproduction, CP has been considered for critically sick COVID-19 patients [54].
In SARS-CoV and MERS, CP was shown to provide NAbs binding to spike1-receptor binding protein (S1-RBD), S1-Nterminal domain and S2, thus blocking entry and containing viral amplification.
Very recently, Cochrane Database of Systematic Reviews published a rapid review on convalescent plasma or hyperimmune immunoglobulin for people with COVID-1940, including 19 studies (2 RCTs, 8 controlled NRSIs, 9 non-controlled NRSIs) with 38,160 participants, of whom 36,081 received convalescent plasma [55]. Two completed RCTs are awaiting assessment (published after 19 August 2020). Were identified a further 138 ongoing studies evaluating convalescent plasma or hyperimmune immunoglobulin, of which 73 are randomised (3 reported in a study registry as already being completed, but without results). No completed studies evaluating hyperimmune immunoglobulin was identified.
The review also includes results from two RCTs (both stopped early) with 189 participants, of whom 95 received convalescent plasma. Control groups received standard care at time of treatment without convalescent plasma.
The conclusions of the review are uncertain whether convalescent plasma decreases all-cause mortality at hospital discharge (risk ratio (RR) 0.55, 95% confidence interval (CI) 0.22 to 1.34; 1 RCT, 86 participants; low-certainty evidence) and whether convalescent plasma decreases mortality (time to event) (hazard ratio (HR) 0.64, 95% CI 0.33 to 1.25; 2 RCTs, 189 participants; low-certainty evidence).
Convalescent plasma may result in little to no difference in improvement of clinical symptoms (i.e. need for respiratory support) at seven days (RR 0.98, 95% CI 0.30 to 3.19; 1 RCT, 103 participants; low-certainty evidence). Convalescent plasma may increase improvement of clinical symptoms at up to 15 days (RR 1.34, 95% CI 0.85 to 2.11; 2 RCTs, 189 participants; low-certainty evidence), and at up to 30 days (RR 1.13, 95% CI 0.88 to 1.43; 2 studies, 188 participants; low-certainty evidence).
No studies reported on quality of life. Reporting of safety data and duration of follow-up was variable. The controlled studies reported on Adverse Events (AEs) and Severe Adverse Events (SAEs) only in participants receiving convalescent plasma. Some, but not all, studies included death as a SAE.
The studies did not report the grade of AEs. Fourteen studies (566 participants) reported on AEs of possible grade 3 or 4 severity. The majority of these AEs were allergic or respiratory events. The studies are very uncertain whether convalescent plasma therapy affects the risk of moderate to severe AEs (very low-certainty evidence).
17 studies (35,944 participants) assessed SAEs for 20,622 of its participants. The majority of participants were from one noncontrolled NRSI (20,000 participants), which reported on SAEs within the first four hours and within an additional seven days after transfusion. There were 63 deaths, 12 were possibly and one was probably related to transfusion. There were 146 SAEs within four hours and 1136 SAEs within seven days post-transfusion. These were predominantly allergic or respiratory, thrombotic or thromboembolic and cardiac events.
A recently published randomized clinical trial published by PlasmAr Study Group has concluded that no significant differences were observed in clinical status or overall mortality between patients treated with convalescent plasma and those who received placebo [56].
All these studies were using not more than 500 ml of convalescent plasma and the scientific reason for the transfusion was the activity of neutralising antibodies against SARS-CoV-2.
In this paper, on the contrary, we have shown that the worsening of Covid-19 clinical symptoms is due to the catastrophic “unfriendly fire” of recruited neutrophils, with overhelming and imbalancing of serine proteolytic cascades activator proteases over inhibitors, serious endotheliopathy, NETosis, ULVWF release, hypercoagulability and diffuse micro/macrothrombi formation.
This condition in the paper has been described as “proteolytic storm”, which advances and sustains/is sustained by the well known “cytokine storm”as shown in Figure: 4.
Following this hypothesis, human plasma, non convalescent, should be used in adequate quantities (more than 2 liters and/or following plasma-exchange procedures)as it supplies:
The SARS-CoV-2 infection is a protease dependent process as cell entry depends on the binding of the Spike protein’s S1 subunit to ACE2 on the target cell surface and host proteases, furin, as well as TMPRSS2 processes the S protein to facilitate membrane fusion, allowing SARS-CoV-2 to have enhanced proteolytic activation in a wider range of tissues.
Aprotinin have been shown to inhibit SARS-CoV-2 entry and replication and thus an aerosol treatment as for influenza is suggested.
After cell entry, SARS-CoV-2 induces a profound immunological unbalance, with a delay of the IFN-I and -III response and upregulation of chemoattractants for neutrophils and monocytes, cytokines storm and increased reactants biomarkers. A PANoptosis process is caused by the combination of TNF-α and IFN-γ
Chemoattractants and cytokines enhance recruitment of neutrophils, while suppressing lymphocytes with an increase of neutrophil/lymphocyte ratio (NLR), which is considered a marker of severe Covid-19. Neutrophils, together with other mononuclear, migrate to the infected alveoli, where they attack SARS-CoV-2, but in addition to direct virus-inflicted pathologies, their exaggerated “unfriendly fire” responses, resulting in a “proteolytic storm” and, inducing alveolar capillary endotheliopathy, contribute to disease severity and subsequent viral dissemination.
One of the mechanisms that neutrophils undertake for host defense is neutrophil extracellular traps (NETs) release, known as NETosis. NETs, via electrostatic interactions, activate the contact pathway of coagulation and, through tissue factor, the intrinsic pathway and form a scaffold for thrombus formation by promoting platelet adhesion and by concentrating coagulation factors involved in clotting.
Concurrently SARS-CoV-2, together with neutrophil elastase and other proteases, induces endothelial cells damage (endotheliopathy) triggering the activation of two independent endothelial pathways and activation of the platelet and endothelial exocytosis of ULVWF (due to ADAMTS13 biosynthesis inhibition), mediating microthrombogenesis and hypercoagulability state. The activation of platelets and clotting proteolytic system involves the other innate serine protease systems with their four major columns, coagulation, fibrinolysis, kallikrein and complement, strictly correlated and interconnected and their physiological maintenance is the result of a rigorous balance between activators and inhibitors. The interplay between complement and coagulation is crucial to understand the clinical implications in Covid-19.
NETs activate Kallikrein/kinin, inducing a prothrombotic state by activating intrinsic pathway and proinflammatory state by producing bioactive peptide bradykinin.
Main family of the serine protease inhibitors (SERPINs) formed by SERPINA1, SERPINA5, SERPINC1, SERPIND, SERPINE1, SERPING1 and SERPINF2 inhibit the major serine protease cascades. Complement activation is inhibited also by Decayaccelerating factor (DAF) and Factor H (alternative pathway). Alpha 2 macroglobulin acts as an antiprotease for a variety of proteases like plasmin, kallikrein and thrombin. A delicate balance between serine proteases and their serpin inhibitors is crucial for normal functioning of biological pathways.
Proteases/antiproteases balance is present also at the endothelial surface (thrombomodulin induces protein C activation to suppress blood coagulation) and also at alveolar space, where SERPINA1 strongly and specifically inhibits neutrophil elastase. When the inhibitor concentration is sufficient to block released elastase, no lesion happens nor in alveolar epithelium nor in alveolar endothelial wall and this corresponds with the moderate Covid-19 clinical condition.
If SERPINA1 is overhelmed or is absent/deficient imbalance between elastase and anti-elastase activity causes progressive damage of both alveoli and endothelium, inducing endotheliopathy and thrombogenic state previously described.
Convalescent plasma has been recently shown uneffective in several clinical trials with no significant differences observed in clinical status or overall mortality between patients treated with convalescent plasma and those who received placebo. All these studies were using not more than 500 ml of convalescent plasma and the scientific reason for the transfusion was the activity of neutralising antibodies against SARS-CoV-2.
In this paper, on the contrary, it has been shown that the worsening of Covid-19 clinical symptoms is due to the catastrophic “unfriendly fire” of recruited neutrophils, with overhelming and imbalancing of serine proteolytic cascades activator proteases over inhibitors, serious endotheliopathy, NETosis, ULVWF release, hypercoagulability and diffuse micro/macrothrombi formation (proteolytic storm).
Thus, the transfusion of human plasma, non convalescent, should be used in adequate quantities (more than 2 liters and/or following plasma-exchange procedures) due to the large quantity of protease inhibitors supplied. Other serine protease inhibitors like aprotinin can be used to restore serine protease balance, working as a “firehose” extinguishing “proteolytic storm”.
SARS-CoV-2 cell entry is a protease dependent process and can be efficiently inhibited by Aprotinin, a serine protease inhibitor.
Severe Covid-19 is the consequence of neutrophils induced “proteolytic storm”, with serine proteases released in large quantities able to overwhelm physiological homeostasis between activators and inhibitors.
Convalescent plasma (CP) transfused to supply NAbs against SARS-CoV-2 in severe Covid-19 patients has failed in several clinical trials showing no significant differences in clinical status or overall mortality between patients treated with CP and those who received placebo and the median volume of infused CP was 500 m.
CP supply NAbs could be useful in the previous phases of SARSCoV-2 infection, but clinical trials with this target are absent.
On the contrary, if CP or human plasma and aprotinin are infused to supply serine protease inhibitors as a “fire extinguisher” for neutrophils ``unfriendly fire” and its consequences of “proteolytic storm”, hypercoagulability, thrombosis and sepsis with MOF, the infused volume should be at least 2000 ml or plasma-exchange should be performed. Clinical trials are needed to confirm this hypothesis.