Author(s): R El Qadiry
Moyamoya disease is a chronic and occlusive cerebro-vascular disease characterized by bilateral steno-occlusive changes in the internal carotid artery and its proximal branches and an abnormal vascular network at the base of the brain. Polycystic Kidney disease (PKD) is known as one of the underlying diseases of moyamoya syndrome which was rarely reported. Only 3 cases of moyamoya syndrome associated with PKD have been reported in the literature. We report here another case of moyamoya syndrome associated with PKD in a Moroccan child.
Case Report: 7-year-old girl, was born of a consanguineous marriage, operated at the age of 7 months for bilateral inguinal hernias with a history of polycystic kidney and liver disease, discovered at D15 of life, complicated by arterial hypertension well-balanced under dual therapy, chronic renal insufficiency under conservative treatment and portal hypertension with hypersplenism and esophageal varices grade II. Admitted for a partial afebrile seizure (left hemibody), tonic-clonic associated with intensive headache. Angio-MRI showed features of Moya-Moya syndrome with amputation of the left sylvian artery, and slender aspect of some portions of the right sylvian artery. Treatment was symptomatic with benign evolution without surgery.
Conclusion: This is the first report of a Moroccan person with PKD associated with moyamoya syndrome.
Moyamoya disease is a chronic and occlusive cerebro-vascular disease characterized by bilateral steno-occlusive changes in the internal carotid artery and its proximal branches and an abnormal vascular network at the base of the brain. The diagnosis of Moyamoya is based upon the characteristic angiographic appearance. Moyamoya disease is considered syndromic when it is associated with other disorders such as neurofibromatosis, trisomy 21 or tuberculous meningitis among others [1].
Polycystic Kidney disease (PKD) is known as one of the underlying diseases of Moyamoya syndrome which was rarely reported. Its pathogenesis remains unknown and its treatment is directed at symptomatic amelioration (Intracranial pressure, cerebral blood flow, and controlling seizures).
Only 3 cases of moyamoya syndrome associated with PKD have been reported in the literature. Here we report the first case of moyamoya syndrome associated with PKD in a Moroccan child.
7-year-old girl was born of a consanguineous marriage with a good psychomotor development, operated at the age of 7 months for bilaterally inguinal hernia. This patient with no family history of kidney disease has been followed for a history of polycystic kidney and liver disease (fig. 1), discovered at 15 day of life on an abdominal ultrasound indicated in front of a perception of a mass on abdominal examination.
During her follow-up, the child had developed multiple complications: arterial hypertension well-balanced under dual therapy (angiotensin converting enzyme (ACE) inhibitors, angiotensin II receptor blocker (ARBs)), chronic renal insufficiency under conservative treatment and portal hypertension with hypersplenism and esophageal varices grade II.
Admitted for a partial seizure (left hemi body), tonic-clonic lasting less than two minutes with a 5-minute post critical motor deficit associated with intensive headache without vomiting or photophobia, all evolving in a context of apyrexia and conservation of general condition.
The clinical examination found, a conscious child, afebrile at 37.3°C, Blood pressure at 98/64 mmHg with normal haemodynamic status. Pupils were isochoric and reactive. She had neither neck stiffness nor motor deficit. She had bilateral bimanually palpable and ballot able masses in the right and the left lumbar region. The rest of the somatic examination was unremarkable.
Cerebral edema of the right hemisphere with mass effect was the main abnormality observed on brain computed tomography. Meanwhile, cerebral MRI showed isosignal intensity in the T1-weighted image and hyper intensities in the T2 and Flairweighted images within the right parietal lobe, low apparent diffusion coefficient (ADC) values due to a recent hypoxicischemic injury and isosignal intensity in the T1-weighted image, hyper intensities in the T2 and Flair-weighted images, with no diffusion abnormalities within the left parietal lobe associated with enlargement of the opposite subarachnoid spaces, along with dilation and gliosis of the opposite crossroads. Angio-MRI showed features of Moya-Moya syndrome with amputation of the left sylvian artery, and slender aspect of some portions of the right sylvian artery (Fig. 2).
Other causes of Moya-Moya syndrome were ruled out (including inflammatory syndromes, thrombophilia, autoimmune diseases, atheroma and arthritis).
Management was symptomatic non-operative focused on reducing intracranial pressure, improving cerebral blood flow, and controlling seizures and headache with favorable evolution.
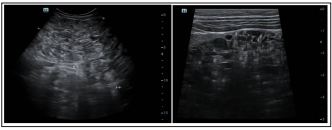
Figure 1: Renal Ultrasound Shows Enlarged Echogenic Kidneys with Multiple Small Cysts Exhibited a Spongiform Appearance
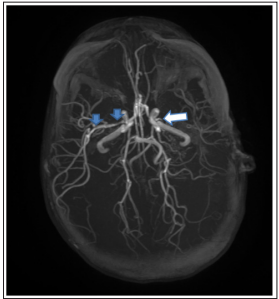
Figure 2: Angio-Mri Showed White Arrow: Amputation of the Left Sylvian Artery.
Blue arrows: Slender Aspect of Some Portions of the Right Sylvian Artery.
The primary lesion in moyamoya disease is stenosis of the intracranial arterial trunk which is thought to be the result of intimal fibrocellular thickening. This leads to the development of the extensive collateral circulation. The annual incidence of moyamoya syndrome is 0.11/100,000, and its prevalence is 4/100,000. The clinical manifestations of Moyamoya are variable and include transient ischemic attack, ischemic stroke, hemorrhagic stroke, and epilepsy [2-4].The etiology of Moyamoya syndrome is largely unknown. However some known risk factors for developing Moyamoya include neurofibromatosis, tuberculous meningitis, connective tissue diseases, irradiation, trisomy 21, cerebral hemorrhage, and cranial trauma [5].
PKD is also known as one of risk factors of moyamoya syndrome which was rarely reported [5]. To our knowledge, only three cases of moyamoya syndrome associated with PKD have been reported in the literature. The first case, reported in 1989 by Massey and Pracyk of patient with a history of multiple cranial traumas, hypertension, polycystic kidney disease, and eosinophilic granuloma [4]. The second case reported by Nzwalo et al. in 2013 of a rare case of moyamoya syndrome in two European Caucasian siblings coexisting with polycystic kidney disease and intestinal duplication cyst [6]. The last case reported in 2020 by Kefang et al. of a 34-year-old patient [3]. We report here the forth case of PKD occurring in association with moyamoya, and the first case in Morocco.
Treatment of Moyamoya syndrome is directed at symptomatic amelioration. The American College of Chest Physicians (ACCP) suggests aspirin as initial therapy for children with acute arterial ischemic stroke secondary to Moyamoya. However, the churgical revascularization is sometimes an option as well [4]. Patients presenting with confounding comorbidities are often difficult to manage. Our patient received symptomatic treatment without aspirin because of the risk of bleeding from esophageal varices. The prognosis is variable.
MMD is a very rare disease. To date, too limited number of related works has been carried out. As such, and because of its rarity, several aspects of this disease remain to be elucidated. The aforementioned case is the first report relating to a Moroccan person suffering from PKD associated with Moyamoya syndrome.
REQ wrote the manuscript, HN I wrote the manuscript with her support, AB supervised the findings of this work, IA supervised the findings of this work.
Informed consent has been obtained from the patient’s parents for publication of the case report.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.