Author(s): Dominique Zöld*, Judit Végh, Júlia Takács and Emese Kiss
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder usually resulting in an increased risk of blood clots in the arterial-, venous-, or microvascular system. Various antibodies have been identified causing the activation and aggregation of thrombocytes and endothelial dysfunction resulting in a wide range of symptoms. Frequent manifestation may be arterial or venous clots in extremities or internal organs, neurological manifestation or pregnancy morbidities. Rarely, APS can manifest in the form of hemorrhage. We discuss the case of a 40-year-old male presenting with a severe headache, fatigue and fever. Head CT confirmed a subdural hematoma. Laboratory findings showed anemia, thrombocytopenia and elongated coagulation times. Immunoserology found the strong presence of lupus anticoagulant, anti-cardiolipin-, and anti-β2glycoprotein-I IgG antibodies confirming the diagnosis of primary antiphospholipid syndrome, specifically LA - hypoprothrombinemia syndrome. Following the administration of high dose glucocorticoids, we chose to give intravenous immunoglobulin followed by rituximab. Although his symptoms regressed and there was no further hemorrhaging, we chose to perform plasmapheresis due to continuously high concentration of anti- phospholipid antibodies to lower the antibody titer, which proved to be effective. Antithrombotic treatment can be challenging in such cases due to the co-occurrence of hypoprothrombinemia and prothrombotic antibodies. We decided to administer low-dose aspirin, with which the patient has no bleeding or blood clots for over 6 years. We find the case worth presenting due to the rare occurance of bleeding as primary manifestation of APS and due to the challenging treatment.
Antiphospholipid syndrome is a systemic autoimmune disorder resulting in an increased risk of blood clots in the arterial-, venous-, or microvascular system. It can occur as a primary disease or secondarily most frequently co-occurring with systemic lupus erythematosus (SLE).
Various antibodies have been identified causing the activation and aggregation of thrombocytes and endothelial dysfunction resulting in thrombotic events or obstetric complications. The diagnostic, most commonly present antibodies target phospholipids or their protein cofactors. Less frequently autoantibodies target e.g. thrombocytes, prothrombin or lead to acquired factor VIII inhibition, resulting in thrombocytopenia, hypoprothrombinemia and possible hemorrhages [1,2].
Symptoms differ widely due to the multiple antibodies causing APS, their heterogeneous mechanism of action and the localisation of the thrombotic clots. Frequent manifestations may be arterial or venous clots in extremities or internal organs, such as the heart or lungs, neurological manifestations or pregnancy morbidities. Catastrophic antiphospholipid syndrome (CAPS) is considered the most severe and often fatal manifestation of the disease developing in approximately 1% of the APS patients where the thrombotic events lead to multiple organ failures over a period of a few days [1,3].
The currently used Sydney classification criteria is based on at least one clinical and one persisting laboratory finding (table 1) [4]. The thrombotic event should be radiologically or pathologically confirmed. Repeated early miscarriages, late intrauterine fetal loss, HELLP syndrome or severe preeclampsia are also considered clinical manifestations.
Table 1: The 2006 Sydney classification criteria for the antiphospholipid syndrome [4]
| Antiphospholipid syndrome (APS) is present if at least one of the clinical criteria and one of the laboratory criteria that follow are met | |
|---|---|
| Clinical criteria | |
| Vacular thrombosis • One or more clinical episodes of arterial, venous, or small vessel thrombosis in any tissue or organ. • Thrombosis must be confirmed by objective validated criteria (i.e. unequivocal findings of appropriate imaging studies or histopathology). • For histopathologic confirmation, thrombosis should be present without significant evidence of inflammation in the vessel wall. |
Pregnancy morbidity a) One or more unexplained deaths of a morphologically normal fetus at or beyond the 10th week of gestation, with normal fetal morphology documented by ultrasound or by direct examination of the fetus or b) One or more premature births of a morphologically normal neonate before the 34th week of gestation because of: eclampsia or severe pre-eclampsia defined according to standard definitions or recognized features of placental insufficiency or (c) Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation, with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes excluded. In studies of populations of patients who have more than one type of pregnancy morbidity, investigators are strongly encouraged to stratify groups of subjects according to a, b, or c above. |
| Laboratory criteria | |
| 1. Lupus anticoagulant present in plasma, on two or more occasions at least 12 weeks apart, detected according to the guidelines of the International Society on Thrombosis and Haemostasis (Scientific Subcommittee on LAs/phospholipiddependent antibodies) 2. Anticardiolipin antibody of IgG and/or IgM isotype in serum or plasma, present in medium or high titer (i.e. >40 GPL or MPL, or >the 99th percentile), on two or more occasions, at least 12 weeks apart, measured by a standardized ELISA. 3. Anti-β2 glycoprotein-I antibody of IgG and/or IgM isotype in serum or plasma (in titer > the 99th percentile), present on two or more occasions, at least 12 weeks apart, measured by a standardized ELISA, according to recommended procedures |
|
The current laboratory criteria require the presence of LA, antiβ2glycoprotein I or anticardiolipin IgM or IgG antibodies in a moderate or high titer over a 12-week period of time [4]. It has been long known that the above antibodies are common in APS, but not present in all patients [5]. It should be noted, that there are several „non-criterion antibodies“ such as IgA serotype anti-CL or anti- β2glycoprotein-I, antiphosphatidylserine or antiprothrombin antibodies etc. that may be present along with or without the criterion antibodies. The latter group has been named seronegative APS (SNAPS). Anti-phosphatidylserine/prothrombin antibodies have been found to be highly effective potential markers for APS [1, 6-9].
The treatment of APS is determined by both the localisation of the clot, as well as additional thrombotic risk factors. Patients with diagnosed systemic autoimmune diseases should be given hydroxychloroquine. Persistent antibody positivity without thrombotic events and low-risk cerebral ischaemic stroke require low-dose aspirin treatment. Other unprovoked arterial or venous thrombotic events should be treated with vitamin K antagonists with a target INR level depending on the risk factors and localisation of the blood clot. For the prevention of obstetric complications low-dose aspirin and prophylactic low molecular weight heparin (LMWH) may be used, where thrombotic events require therapeutic dosage of LMWH with anti-Xa monitoring. CAPS demands a multimodal and a combined therapy of anticoagulant and anti-inflammatory treatment (corticosteroids, plasmapheresis, IVIg, rituximab). In case of failure of standard treatment, several additional treatment options can be considered. [1,3,10,11].
We present the case of a 40-year-old male who had no previous history of any internal medical disease. In December 2015 the patient presented with a progressive headache. The head CT scan showed a subdural hematoma (Image 1). As the neurosurgical consult found no need for intervention, the patient was admitted to the Neurological Department of the Flór Ferenc Hospital. On the third day of treatment he suddenly became somnolent and anisocoric. Due to worsening neurological symptoms and the growth of the haematoma on the control CT scan, the neurosurgical decision was made to perform the evacuation of the hematoma. Laboratory findings already then showed elongated coagulation times, suggesting abnormal hemostasis.
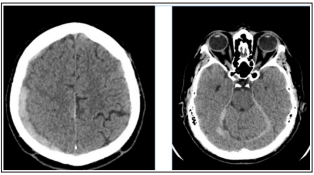
Image 1: Head CT showing parietal, falx and tentorial subdural haematoma
A few weeks later the patient presented with unbearable headache and nausea. The head CT scan found a fresh subdural hematoma (50x11mm) on the right side of the frontal lobe. Due to the progressing symptoms a control CT was once again performed showing minimal progression. Neurosurgical consult found no need for intervention in either cases. Conservative treatment was given in form of dehydration. Throughout the treatment the patient developed a fever and was transferred to the Neurological Clinic of Semmelweis University. Laboratory findings showed a mildly elevated CRP levels with negative PCT. Due to elongated coagulation times further tests were performed. The strong presence of lupus anticoagulant raised the possibility of systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS). The patient was transferred to the Intensive Care Unit of our department. Upon admission, he was fatigued and complained of an unbearable headache. Laboratory findings showed anemia, thrombocytopenia, hypoprothrombinemia, elongated prothrombin time (PT) and activated partial thromboplastin time (aPTT) as well as elevated CRP and Westergren. PCT was within normal range, and the hemoculture was negative. Infection was ruled out. Besides the known LA positivity, the immunoserology proved strong anti-cardiolipin-, anti-β2glycoprotein-I IgG antibody and atypical ANCA positivity along with hypocomplementemia. The elongated prothrombin time raised the possibility of anti-prothrombin antibody, which we were unable to test at the time. Concluding the imunoserological, laboratory findings along with the clinical state, we diagnosed primary antiphospholipid syndrome, specifically LA - hypoprothrombinemia syndrome. The head MRI showed no new lesions (Image 2.). Although all three diagnostic antiphospholipid antibodies were extremely high, we chose to avoid plasmapheresis due to anemia, thrombocytopenia, elongated coagulation times and hypocomplementemia and started the patient on high dose glucocorticoids along with intravenous immunoglobulin (1g/ kg = 80g over 4 days) while continuing dehydration in form of mannitol and dexamethasone. On the third day of treatment the patient was no longer febrile, his headache eased and he started regaining his appetite and strength. Simultaneously the elevated inflammatory markers regressed, the hemoglobin level entered the normal range, coagulation times improved, head CT showed regression and the glucocorticoid dose could be tapered to 1mg / kg. We considered anticoagulation following the improvement of coagulation times due to the high levels of all three diagnostic antiphospholipid antibodies. Taking into account the risk of hemorrhage we decided to avoid vitamin K antagonists and administer low- dose aspirin. The patient was then started on methotrexate and chloroquine adjusted to his weight. Although his general state showed improvement, the immunoserological control still showed highly elevated levels of all three diagnostic APS autoantibodies, indicating the need to intensify treatment. We decided to administer rituximab two times per half a year in a dose of 1000mg with 2 weeks difference. The patient’s symptoms and immunoserological activity regressed and glucocorticoid treatment could be discontinued. Due to gastroenterological side effects methotrexate was stopped after a few months.
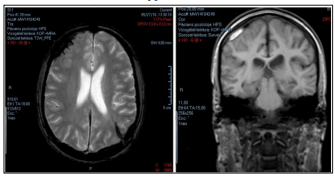
Image 2: Control MRI showing previous laesions
In 2017 the patient presented once again with fever and a severe headache. Physical examination showed the slight oedema of the right maxillary area. Head CT proved no new hemorrhage, however found a right maxillary sinusitis which resolved with antibiotics. Following the third cycle of rituximab the immunoserological results still showed highly elevated LA, a-CL IgG and antiβ2glycoprotein I IgG levels. Although the patient had no new neurological symptoms the treatment option was made to perform plasmapheresis 4 times over a period of 8 days prior to giving the fourth cycle of rituximab due to the continuously high antiphospholipid antibody titers.
The patient was thereafter stable continuing the low-dose aspirin, chloroquine, statin and vitamin-D treatment.
Our patient presented with a subdural hematoma, which is an infrequent primary manifestation of APS. The repeatedly high levels of criterion antiphospholipid antibodies is generally associated with an increased risk of thrombotic events [12]. We find it noteworthy due to the unusual manifestation of hemorrhage despite the continuous high elevation of all three diagnostic antibody titers. APS coagulopathy can manifest in form of haemorrhagic syndrome. Possible causes may be severe thrombocytopenia or an antibody mediated acquired factor VIII or factor II (prothrombin) deficiency. Antiphospholipid mediated thrombocytopenia is usually moderate and rarely the cause of severe hemorrhage in APS [13].
Several case reports have been published where hemorrhage occurs in the presence of LA – namely LA - hypoprothrombinemia syndrome (LAHS) [14]. LAHS is characterised by hypoprothrombinemia in the presence of LA caused by antiprothrombin antibodies. It is considered more frequent in children or females with SLE [14]. Although we were unable to perform the test for antiprothrombin antibody, considering the elongated coagulation times, hypoprothrombinemia, normal factor VIII activity, mild thrombocytopaenia and diagnosis of APS, it seems likely to have been present. As found in our patient, laboratory findings such as prolonged PT, APTT and INR as well as positive LA laboratory findings raise the suspicion of LAHS [15].
We are currently unaware of standardised treatment for LAHS. Several case reports have achieved results using glucocorticoids and cyclophosphamide. As glucocorticoids did not give sufficient results, we chose to administer IVIg followed by rituximab and plasmapheresis to lower antiphospholipid antibody titers, which proved to be effective. The decision of anticoagulation in such cases is difficult. Due to the repeated and growing intracranial hemorrhages we decided to administer low-dose aspirin, with which the patient had no further bleeding or blood clots for over 6 years.
Our 40-year-old patient’s primary manifestation of APS occurred as a subdural hemorrhage. Considering his highly elevated antiphospholipid antibodies, elevated inflammatory markers and hypoprothrombinemia we chose to administer glucocorticoids, IVIg, rituximab, low-dose aspirin, chloroquine, rosuvastatin and vitamin-D. We were able to achieve remission and lower his antiphospholipid antibody titers. To our knowledge he has had no further relapses since. Treatment can be challenging due to the coocurance of hypoprothrombinemia and prothrombotic antibodies.