Author(s): Nisha and Vijay Paramanik*
ABSTRACT
Autophagy related gene-5 (ATG5) acts as a marker for autophagosome formation and initiation of autophagy flux. For performing these functions, ATG5 may interacts with several proteins. Hence, this study aims to identify ATG5 interacting proteins and their regulatory features using in silico approaches. Briefly, Search Tool for the retrieval of interacting genes/proteins (STRING) database predicted thirty-four ATG5 interacting proteins. Physicochemical characterization of ATG5 interacting proteins were predicted using expert protein analysis system (ExPASY) Protparam. Out of thirty-four proteins, five were stable with acidic PI and hydrophilic in nature. The secondary structure and amino acid (aa) content were predicted by Self-Optimized Prediction Method from Alignment (SOPMA). ATG5 interacting proteins comprised with α-helix and random coils. In addition, ATG5 interacting proteins were rich in leucine and serine whereas cysteine and histidine were very few. Functional characterization by Secondary Structure Prediction of Membrane Proteins (SOSUI) database predicted five soluble signal peptides and two membrane proteins. Further, motifs of ATG5 interacting proteins were predicted by MOTIF search. Herein, motifs were predicted in prosite pattern, prosite profile and protein family database (pFAM) category. In this study, active sites of ATG5 interacting proteins were predicted by universal protein resource (UniProt) database showing glycyl thioester intermediate sites maximally. In conclusion, predicting different properties and binding sites may contribute a better understanding of ATG5 regulatory functions and protein-protein interactions that may give potential target for docking studies.
Autophagy is essential to maintain organism’s homeostasis by regulating basic metabolic operations inside cells to diseases like neurodegenerative disorders and lysosomal disorders etc. However, aggregated cargoes, dysfunctional organelles, random cytoplasmic proteins and stored nutrients are controlled by autophagy [1]. Autophagy related gene-5 (ATG5) shows significant role in the development of autophagosome structure that after engulfing degraded proteins fuses with lysosomes to form autophagolysosomal complex for further recycling [2,3].
ATG5 contains 275 amino acids, located on the chromosome 10 in mouse. It acts after conjugating with autophagy-related protein 12 (ATG12) and autophagy-related protein 16l-1 (ATG16l1) forming ATG5-ATG12-ATG16l1 complex. ATG5 shows crucial role in central nervous system (CNS) development and neuronal plasticity showed that crossing conditional ATG5 knockout mice lead to axonal swelling especially in terminal membrane regions [4,5]. Therefore, ATG5 may show role in the regulation of membrane structures near axons. Several computational methods have been used to determine membrane dynamics and predict their functions. In autophagy, the routine clearance of degraded proteins from neuronal cells is an essential step to prevent proteotoxicity.
Protein-protein interaction (PPI) network help us to analyze various molecular function, biological processes and cellular components [6]. In molecular function proteins catalytic activity can be determined. ATG5 induces catalysis by reorienting cysteine-threonine residue of autophagy-related protein 3 (ATG3) conjugate activity. In biological function ATG5 cellular signaling mechanisms and metabolic pathway can be determined. Further, intracellular components show the compartment of the cells where proteins function. These components can be predicted by different methods that predicts signal sequences, membrane association or post- translational modifications. Therefore, various computational tools like MOTIF search, SOSUI, SOPMA etc were used to predict these parameters.
During depression, changes in the energy conversion caused by reduced degradation rate of specific proteins are observed. This may be due to blocking of energy metabolism by mitochondrial autophagy process. In stress conditions, ATG5 maintains mitochondrial membrane potential [7]. A newborn mouse without ATG5 dies within a day due to energy reduction [8]. This shows the efficient relevance of autophagy.
These proofs systemic ablation of core autophagy genes might lead to embryonic or perinatal lethality [9].
Thus, the present study focuses in silico prediction of ATG5 interacting proteins as well as their physicochemical, structural and functional characteristics. Predicted structural and molecular function of ATG5 interacting proteins sequence were retrieved using STRING server. ExPASY database was used to predict physicochemical properties namely molecular weight, isoelectric point (pI), extinction coefficient, instability index, protein charges, aliphatic index and grand average of hydropathy (GRAVY), while SOPMA server was used to predict functional properties like secondary structure and MOTIF search and InterPro database was used to identify motifs and active sites respectively. In silico analysis therefore helpful to develop pharmacokinetically safe and stable molecules. Moreover, it may give potential target for docking studies.
The protein sequence for Mus musculus ATG5 was retrieved using STRING database( https://string-db.org/ ) and confirmed from National Centre for Biotechnology Information (NCBI) (Protein ID: NP_444299) STRING database is an in-memory database which is used for cataloging protein sequences and associated information. STRING is a database that keeps track of known or expected protein interactions based on four different sources: genomics, high-throughput studies, conserved co-expression and previously published literature.
Bioinformatics relies largely on physicochemical properties, especially for analysing and predicting biological macromolecules like proteins and nucleic acids. These characteristics arise from the molecular and physical qualities of nucleotide bases (for nucleic acids) or amino acids (for proteins). The physical and chemical attributes, such as molecular weight, theoretical pI, amino acid composition, extinction coefficient, instability index, aliphatic index and grand average of hydropathy (GRAVY) of the ATG5 proteins were computed using ExPASY ProtParam tool (http:// web.expasy.org/protparam/).
The secondary structure is very popular feature used to encode structure information of amino acids in PPI site prediction. The secondary structure properties like the α-helix, β-turn, extended strand, random coil and turn of amino acid sequences of ATG5 proteins were predicted using self-optimized prediction method with alignment (SOPMA) in percentage ( https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html ).
In bioinformatics, functional characterisation of proteins involves identifying and understanding the functions, relationships, and biological activities of proteins. Since proteins are essential to many cellular functions, study biological systems require an understanding of this process. ATG5 interacting proteins functional characterization like transmembrane region and their solubility were predicted using SOSUI server ( https://harrier.nagahama-i-bio.ac.jp/sosui/mobile/ ).
Motif search is a synchronised web resource for identification of protein motifs by PROSITE and families by using pFAM for investigating their interactions ( https://www.genome.jp/tools/motif/ ). The MOTIF search database includes a range of analysis and visualisation tools in addition to manually selected hidden Markov models for many different fields [10].
A crucial component of structural bioinformatics and bioinformatics in general is the prediction of interacting proteins’ active sites. Finding these areas is useful for drug discovery, enzyme engineering, and functional annotation as well as for understanding the molecular mechanisms driving protein-protein interactions. ATG5 interacting proteins active sites were predicted using InterPro database. Prediction of active sites gives more idea for protein-protein interaction studies.
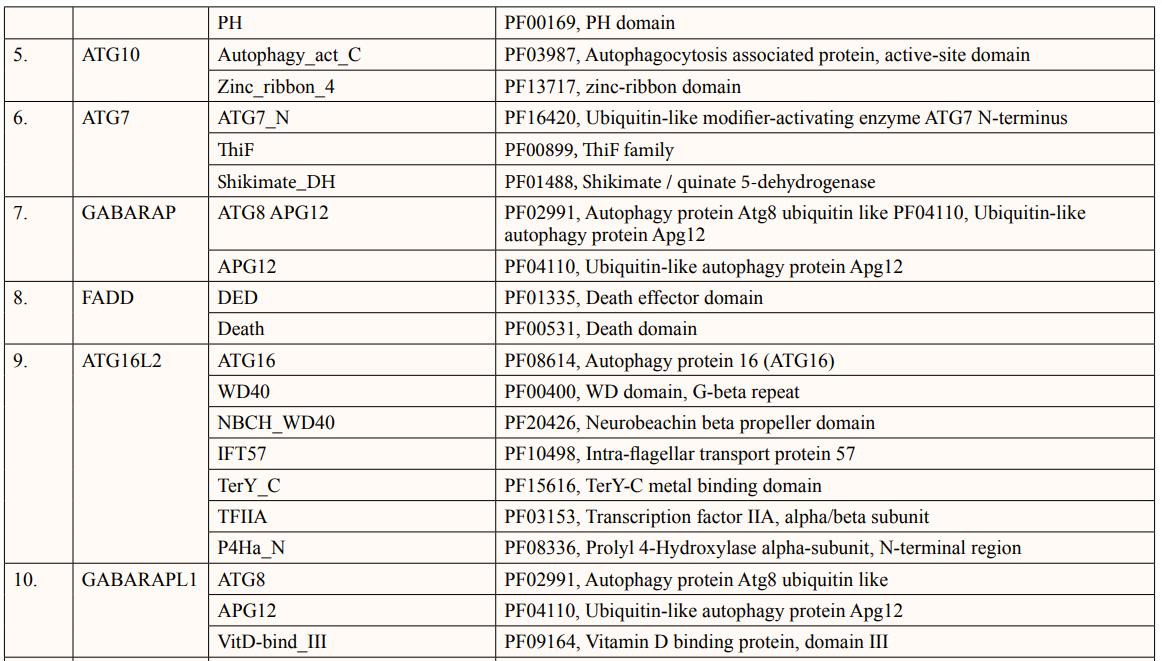
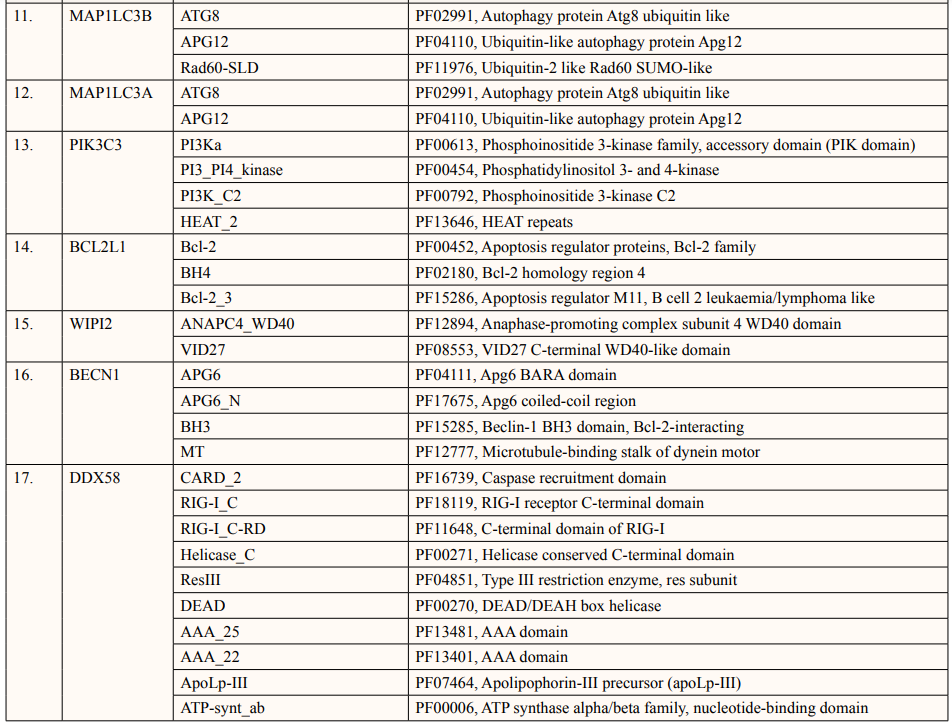
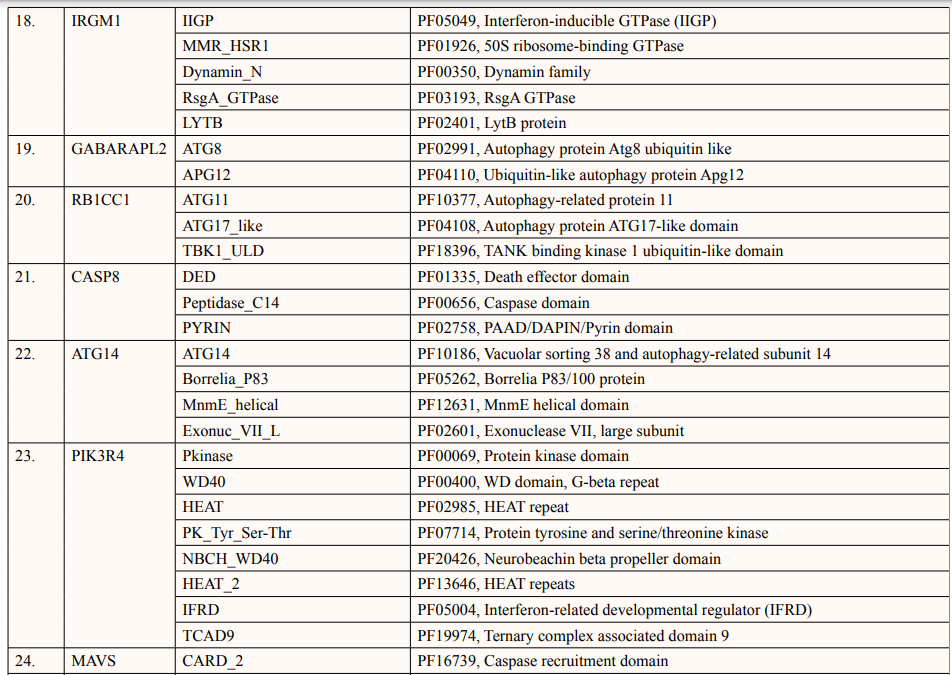
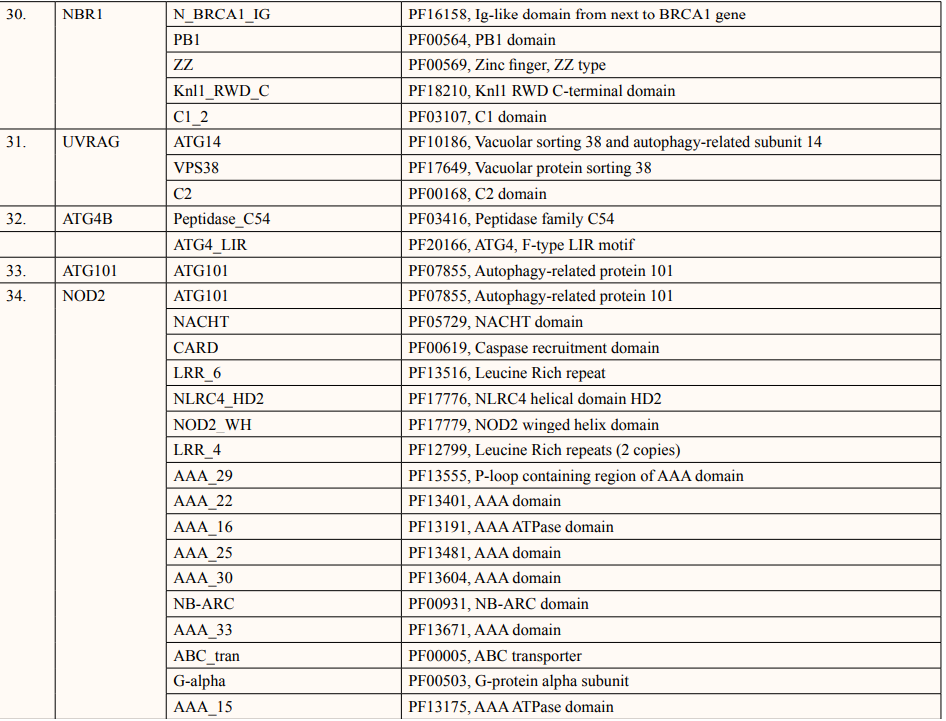
A list of thirty-four ATG5 interactive proteins were found as mentioned in Table 1 and Figure. 1, based on the STRING server. These databases provide information on proteins identified in primary protein databases such as PDB, SWISS PROT and others [11].
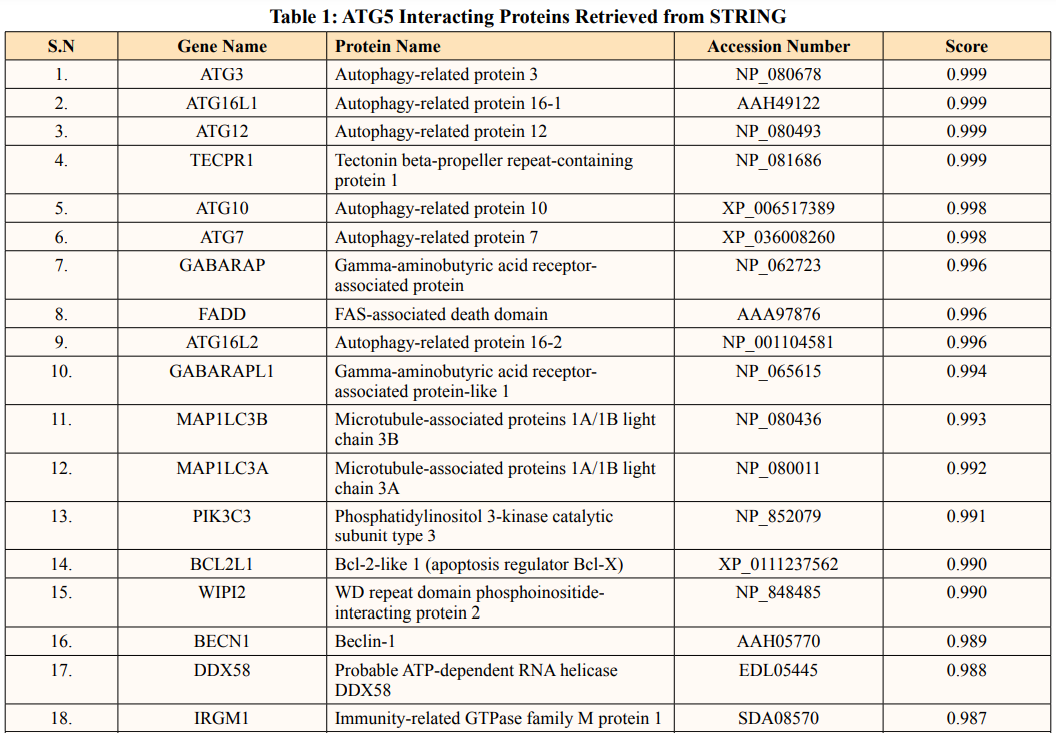
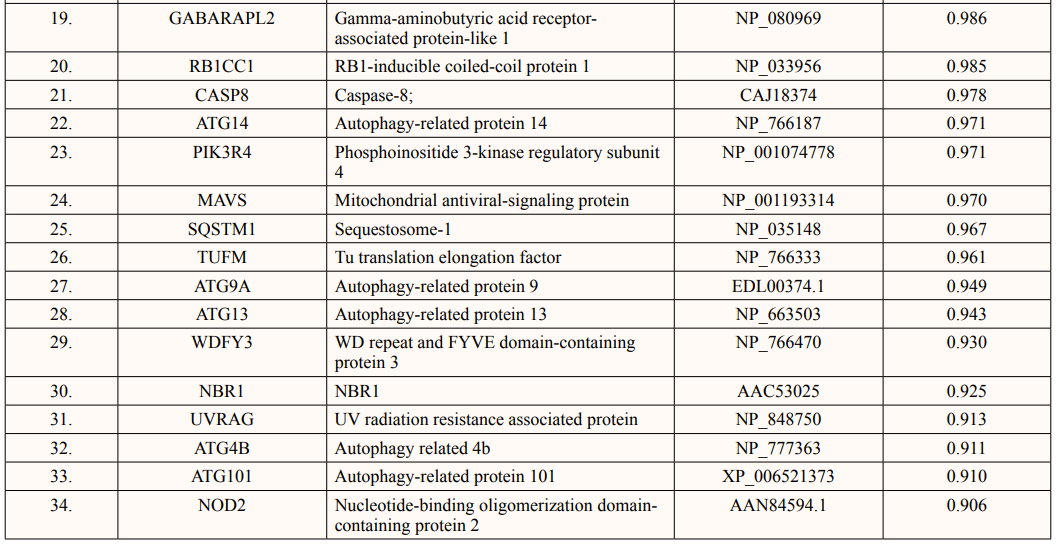
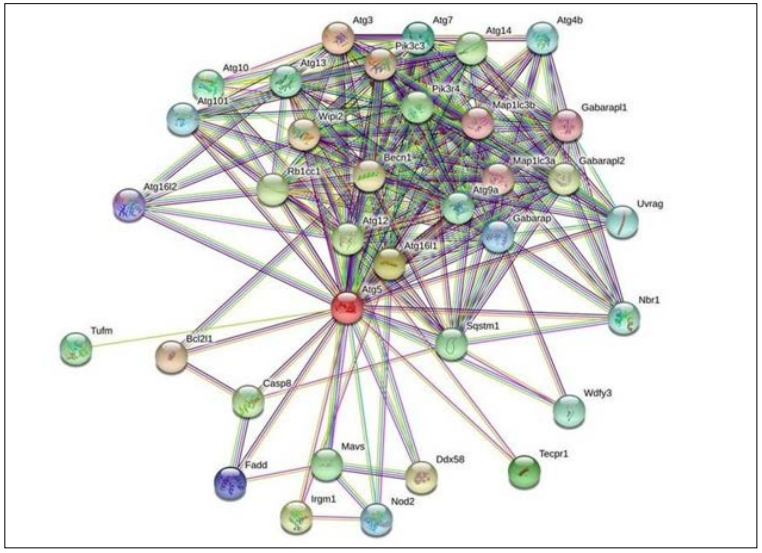
Figure 1: The Figure Indicates STRING Network view for ATG5 Interacting proteins. Coloured Nodes represent Query Proteins and First Shell of Interaction, Filled Nodes Represent some 3D Structure is known and Predicted. Coloured lines between Proteins Indicated Various types of Interactions
Theoretical pI (isoelectric point) of a protein sequence is largely determined by amino acid composition of proteins (Table 2), which is based on a combination of dissociation constant (pKa) values of the constituent amino acids. pI values have often been used in protein isolation, separation, purification, crystallization and other operations to distinguish between proteins and to create various buffer systems [12,13]. Here twenty-four ATG5 interacting proteins namely: ATG3, ATG16l1, autophagy-related protein 12 (ATG12), tectonin beta-propeller repeat-containing protein 1 (TECPR1), autophagy-related protein 10 (ATG10), autophagy-related protein 7 (ATG7), FAS-associated death domain (FADD), autophagy-related protein 101 (ATG101), autophagy-related protein 13 (ATG13), autophagy-related protein 4B (ATG4B), autophagy-related protein 9A (ATG9A), Bcl-2-like 1 (apoptosis regulator Bcl-X) (BCL2L1), Beclin-1 (BECN1), Caspase-8 (CASP8), Probable ATP-dependent RNA helicase DDX58 (DDX58), mitochondrial antiviral-signaling protein (MAVS), (NBR1), phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), phosphatidylinositol 3-kinase catalytic subunit type 4 (PIK3R4), RB1-inducible coiled-coil protein 1 (RB1CC1), Sequestosome-1 (SQSTM1), WD repeat and FYVE domain-containing protein 3 (WDFY3), WD repeat domain phosphoinositide-interacting protein 2 (WIPI2) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2) showed value lower than 7 indicating their acidic nature and other ten proteins i.e., Gamma-aminobutyric acid receptor-associated protein (GABARAP), autophagy-related protein 16l-2 (ATG16L2), gamma-aminobutyric acid receptor-associated protein-Like 1 (GABARAPL1), gamma- aminobutyric acid receptor- associated protein-Like 2 (GABARAPL2), autophagy-related protein 14 (ATG14), immunity-related GTPase family M protein 1 (IRGM1), microtubule- associated proteins 1A/1B light chain 3A (MAP1LC3A), microtubule-associated proteins 1A/1B light chain 3B (MAP1LC3B), Tu translation elongation factor (TUFM), and UV radiation resistance associated protein (UVRAG) had pI value greater than 7 showing basic nature (Table 2).
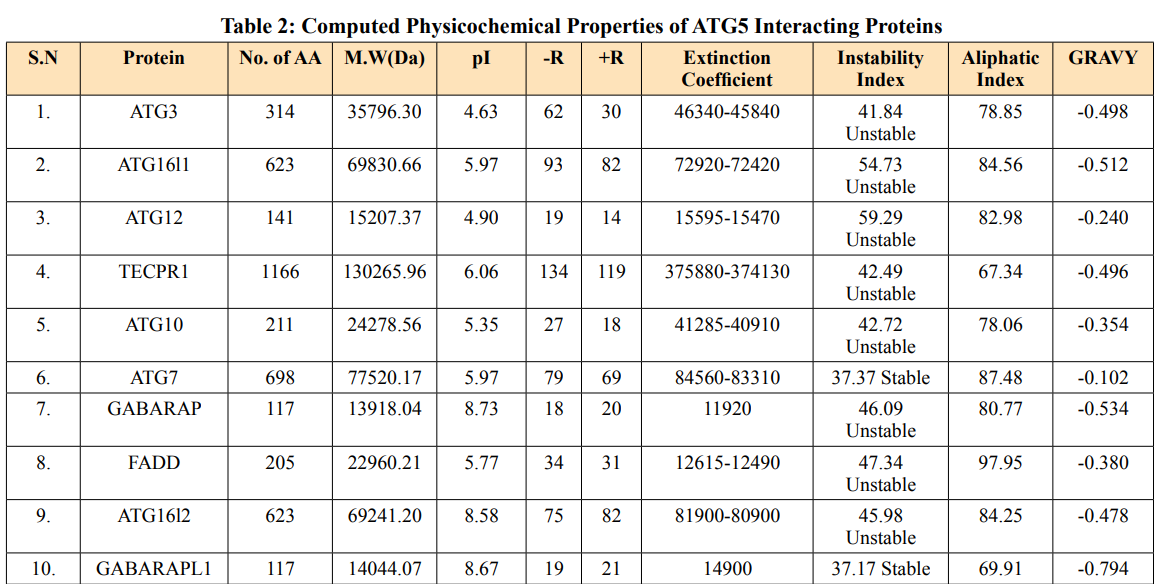
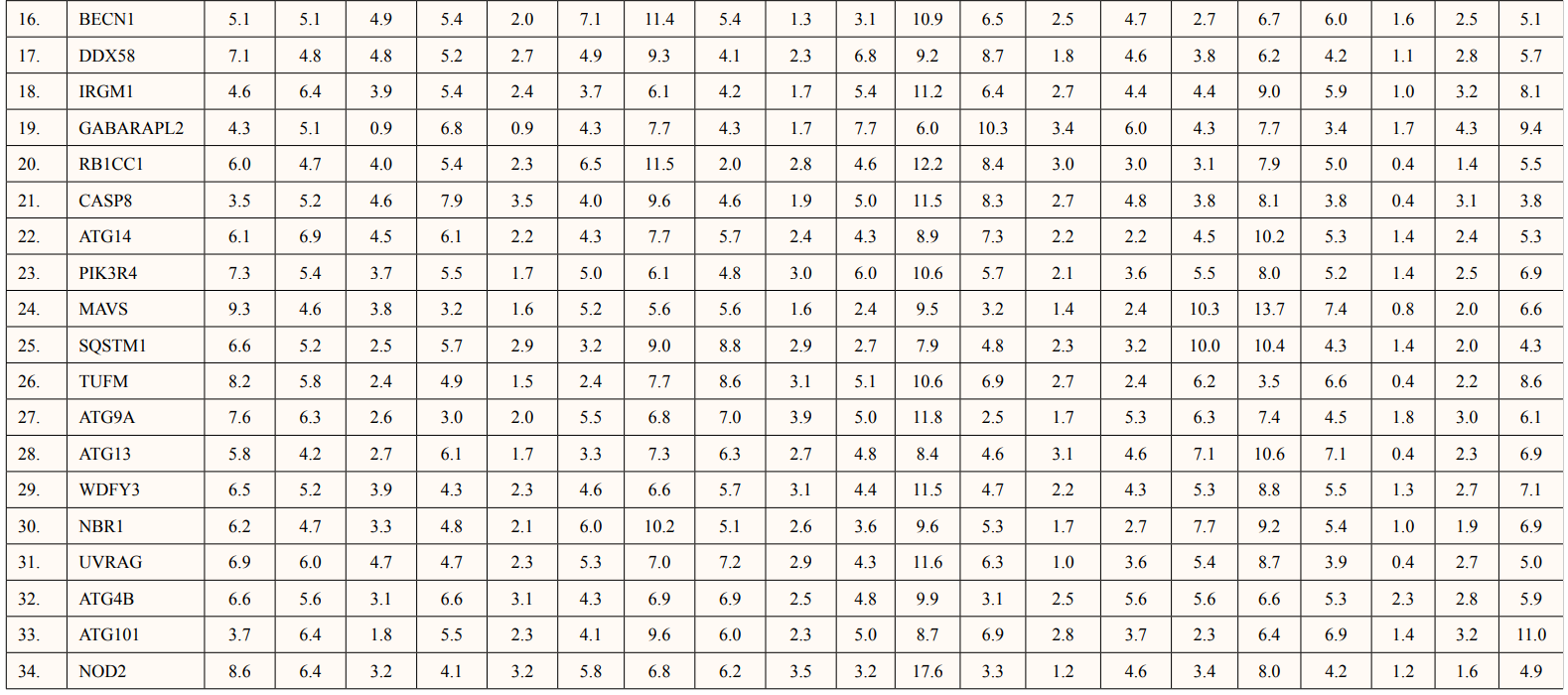

Extinction coefficients are measured in units of M-1 cm-1, at 280nm measured in water [14]. The calculated EC is in direct correlation with the cysteine, tryptophan and tyrosine content of ATG5 interacting proteins. WDFY3 showed highest extinction coefficient among ATG5 interacting proteins in range of 387060-382060 M-1 cm-1. Two proteins viz.MAP1LC3A and MAP1LC3B showed low EC value of 5960 M-1 cm-1 each (Table 2).
The instability index is a way for determining whether a protein will remain stable under various cellular conditions. If instability index is lower than forty, then proteins are likely to remain stable and instability index higher than forty predicts unstable [15]. ExPASY tool predicted that the instability index of ATG5 interacting proteins showed five proteins namely: ATG7, GABARRAPL1, GABARAPL2, BCL2L1 and WIPI2 were stable in nature and remaining twenty-nine proteins namely: ATG3, ATG16L1, ATG12, TECPR1, ATG10, GABARAP, FADD, ATG16L2, ATG101, ATG12, ATG14, ATG4B, ATG9A, BECN1, CASP8, DDX58, IRGM1, MAP1LC3A, MAP1LC3B, MAVS, NBR1, PIK3C3, PIK3C4, RB1CC1, SQSTM1, TUFM, UVRAG, WDFY3 and NOD2 were unstable (Table 2).
The relative volume filled by aliphatic side chains (alanine, valine, isoleucine and leucine) is defined as the aliphatic index of a protein [16]. It could be interpreted as a beneficial factor in favor of protein thermostability. Aliphatic index of ATG5 interacting proteins ranged from 60.50 to 103.90. ATG5 interacting protein SQSTM1 and Tecpr1 showed low thermal stability of 60.50 and 67.34 respectively. In contrast, NOD2 and FADD showed higher aliphatic index of 103.90 and 97.95 respectively. The remaining thirty interacting proteins ranged from 69.91 to 94.47 (Table 2).
The total hydropathy values of all the amino acids in a protein is divided by the number of aa residues to get the GRAVY value [17]. ATG5 interacting proteins showed negative GRAVY values which predicts its non-polar nature except NOD2 protein having positive GRAVY value of 0.009 indicating its polar nature (Table 2).
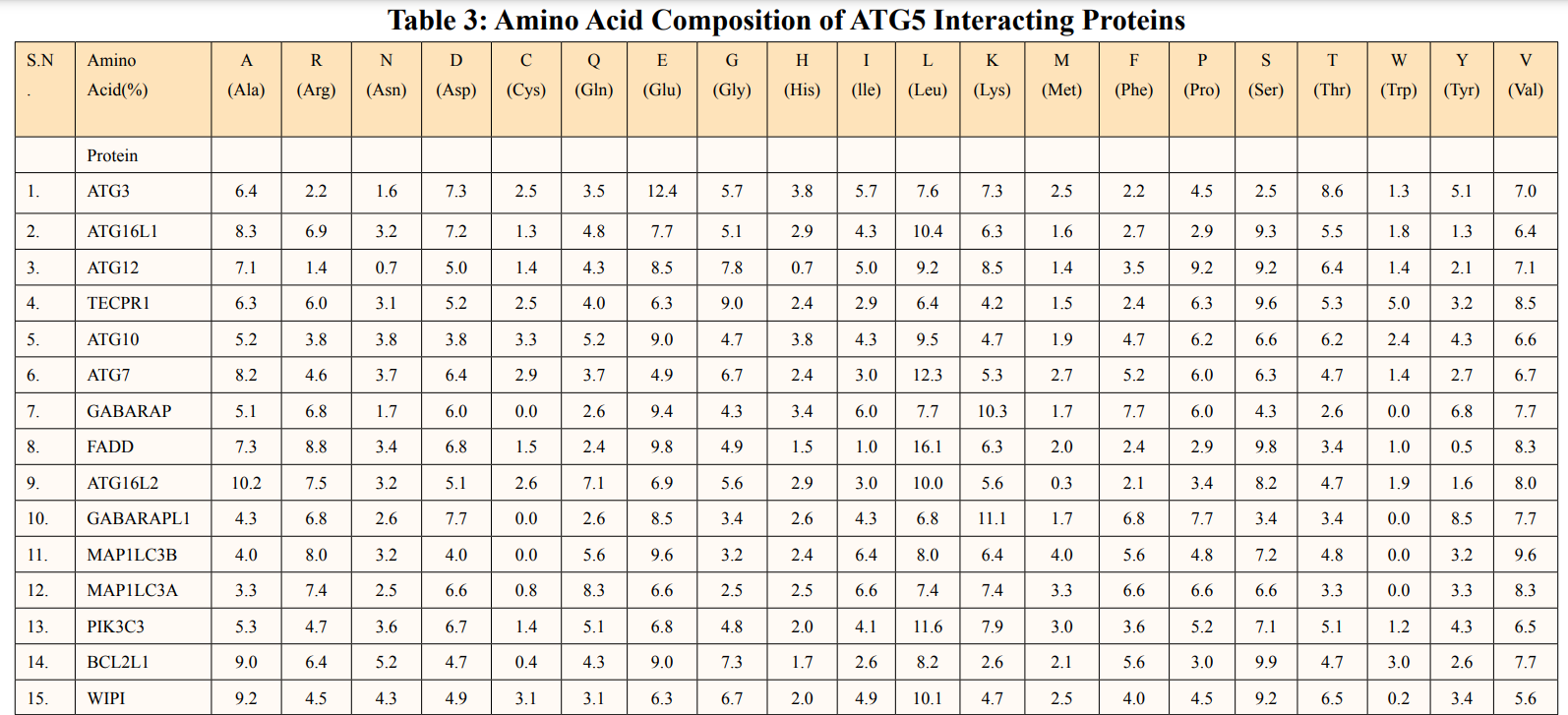
Amino acid composition of ATG5 interacting proteins were given in Table 3. ATG5 interacting proteins contain higher percentage of hydrophobic amino acid leucine and neutral amino acid serine. Also, low percentage of cysteine and histidine amino acids were predicted.
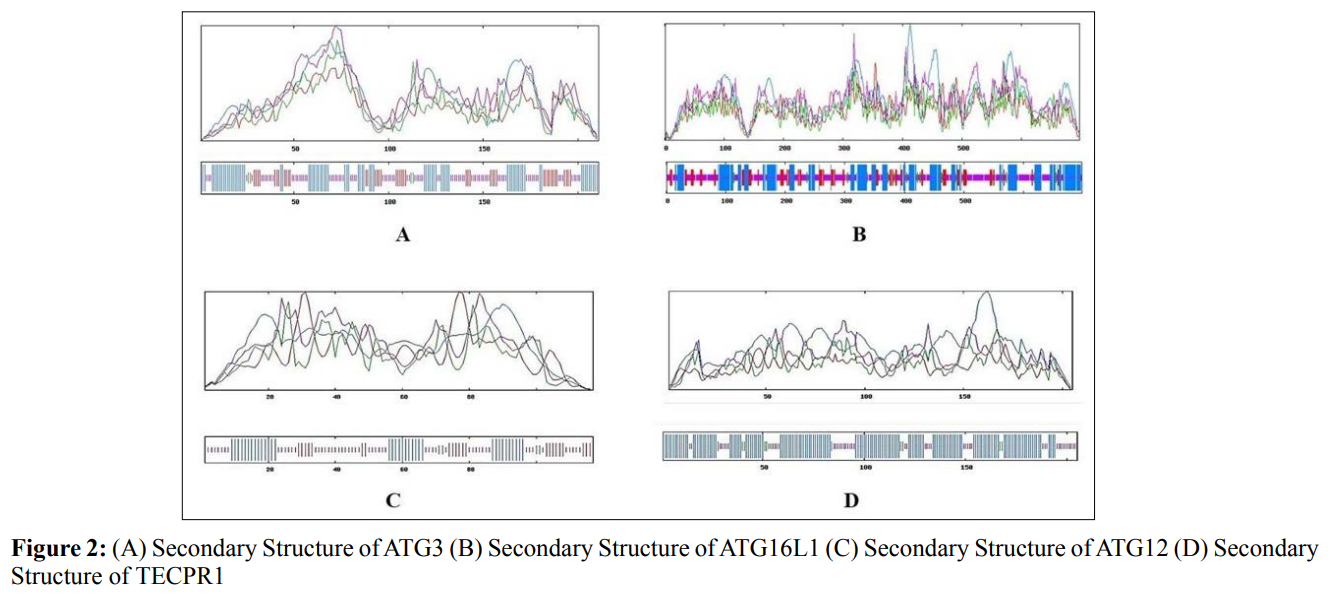
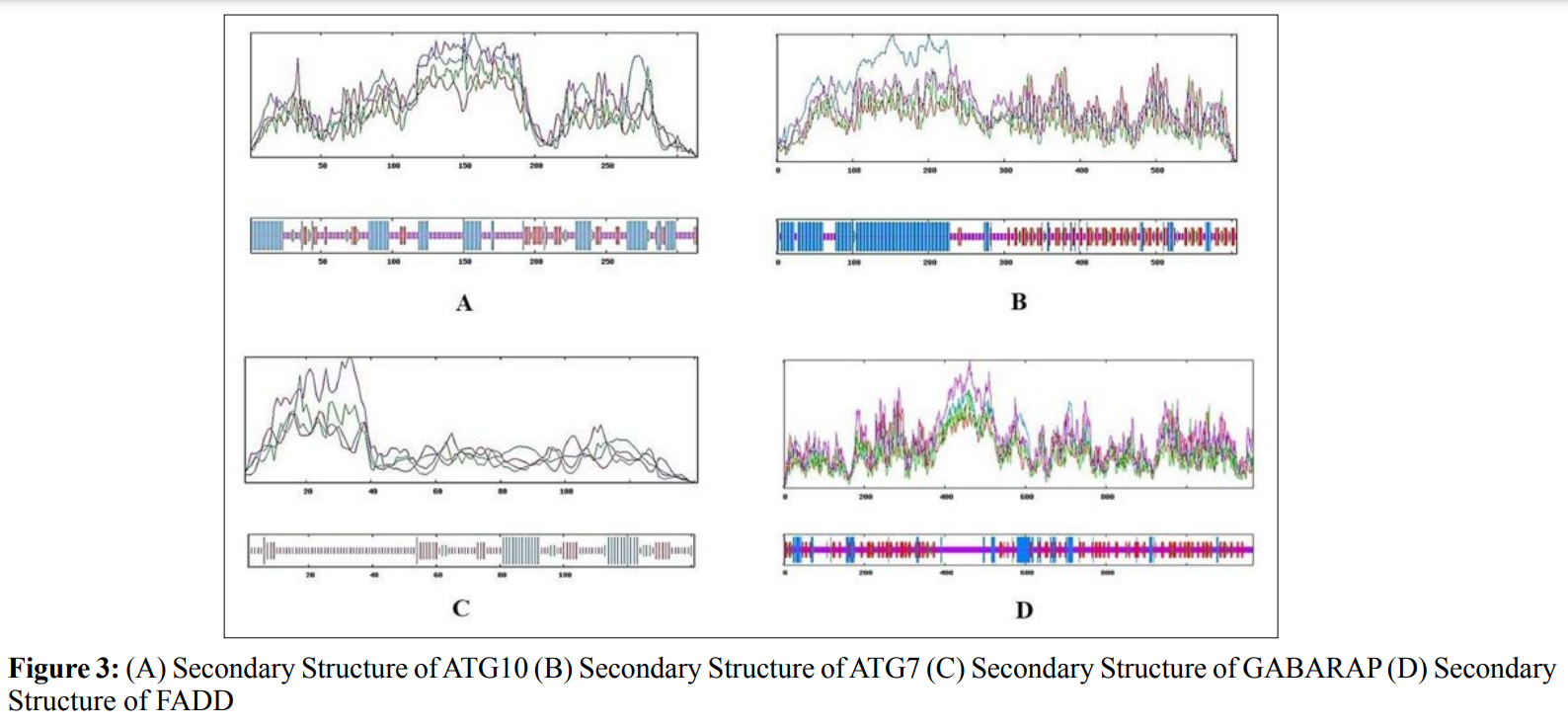
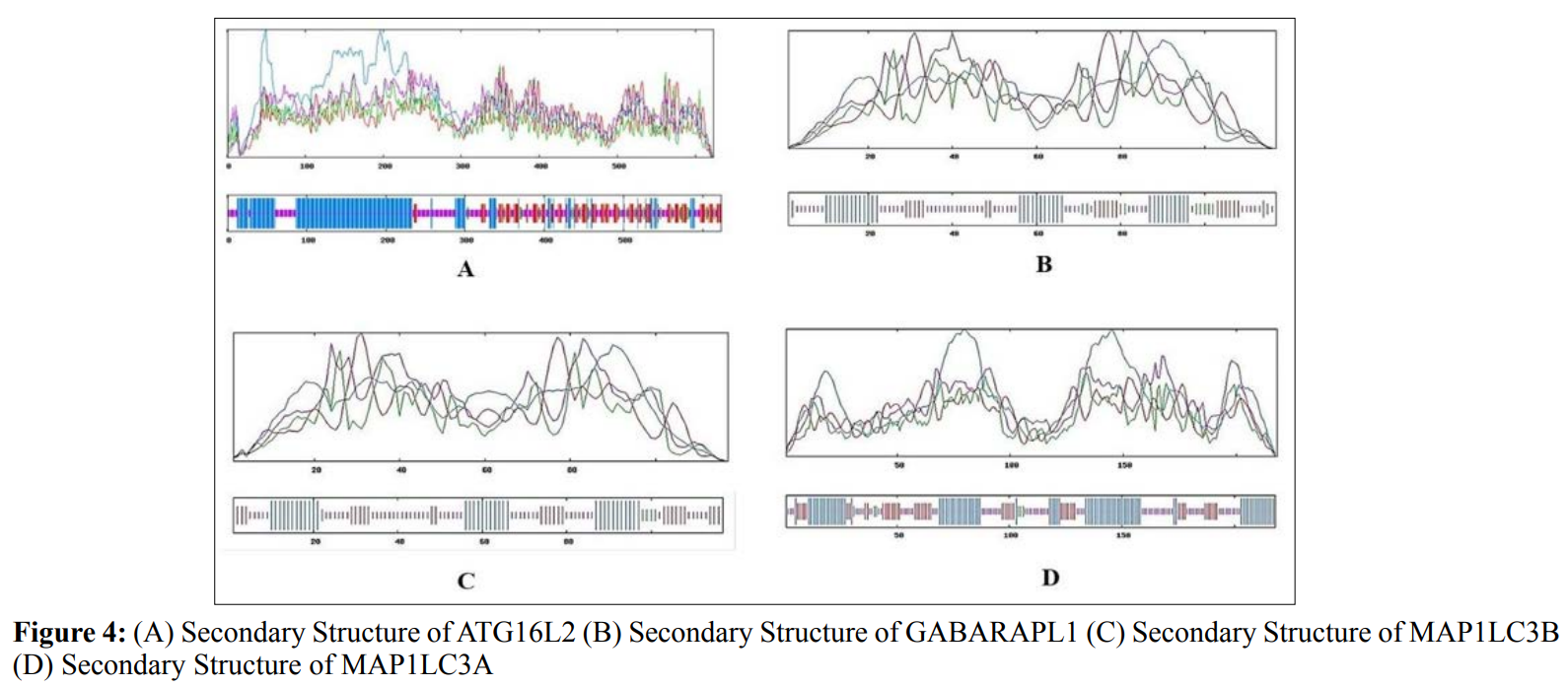
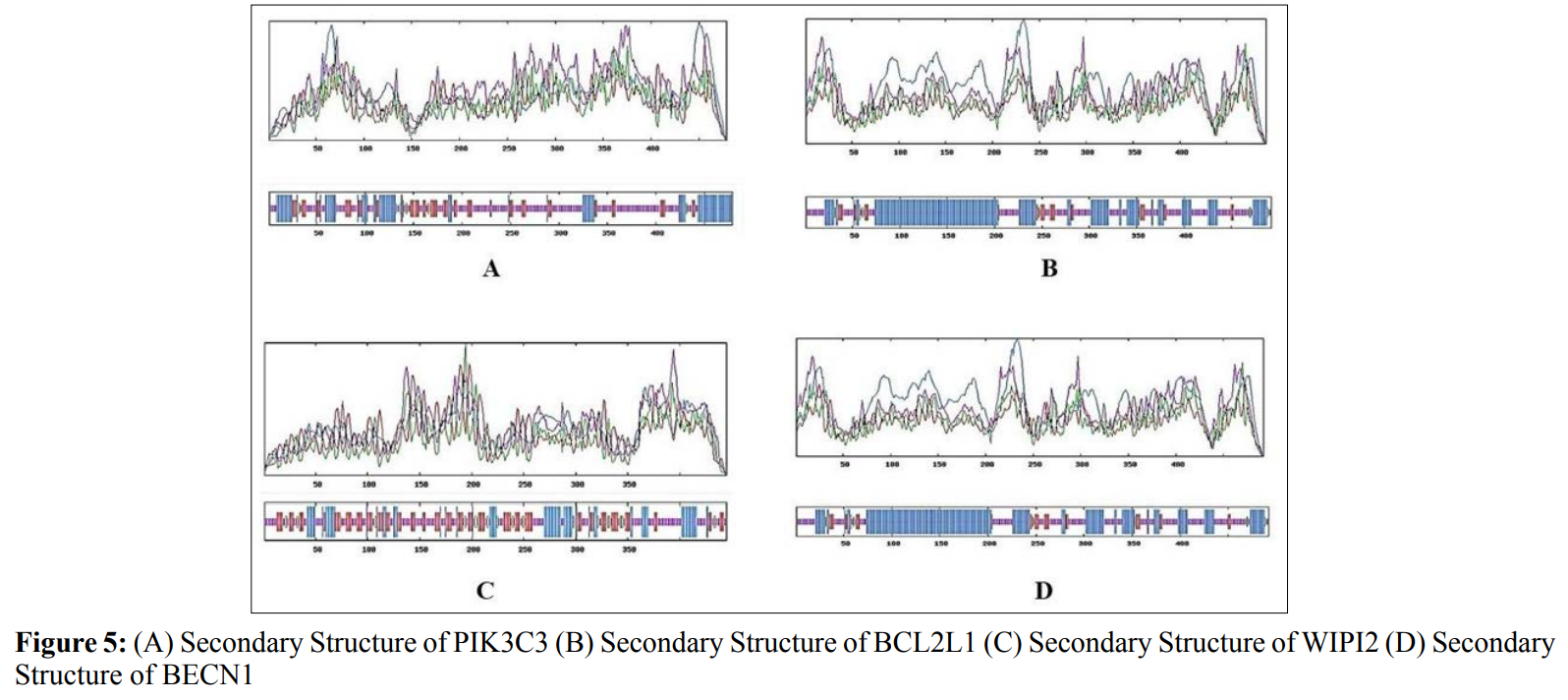
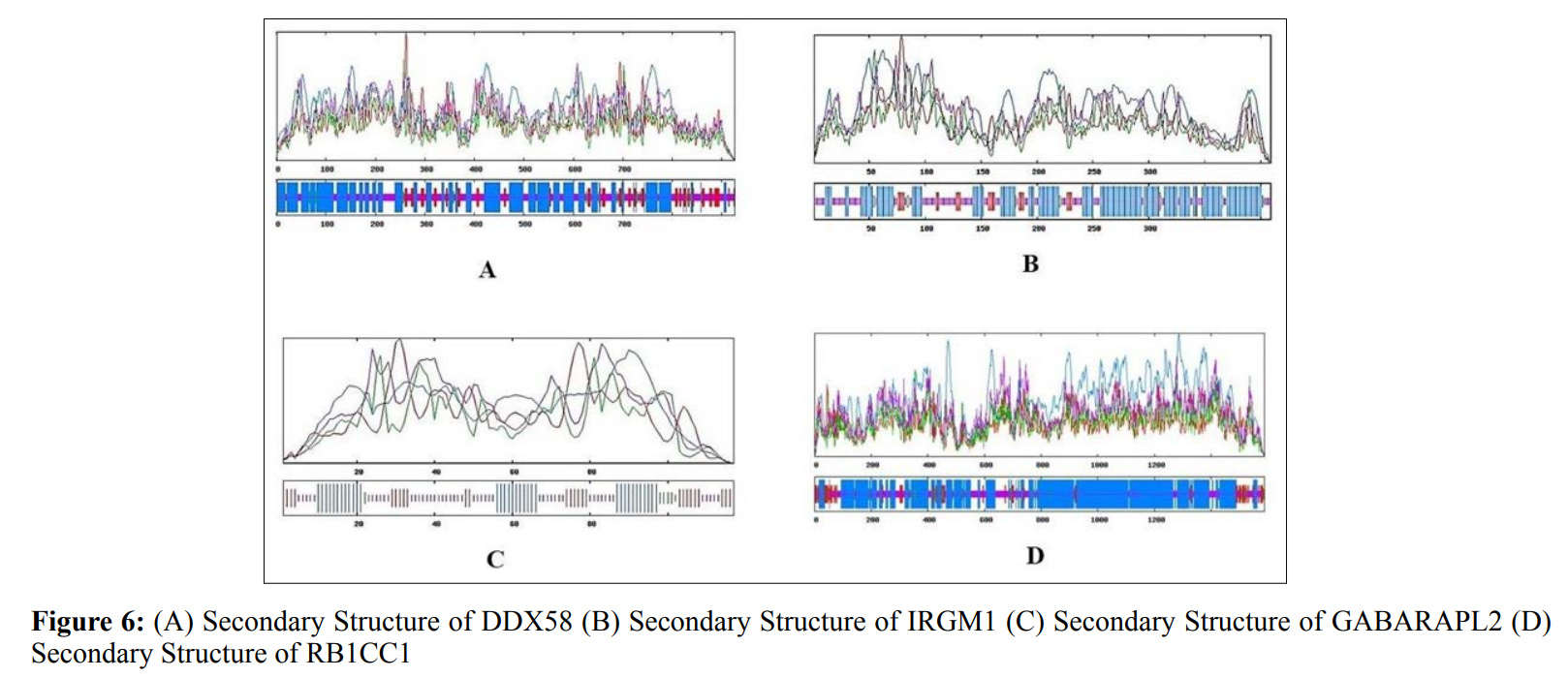
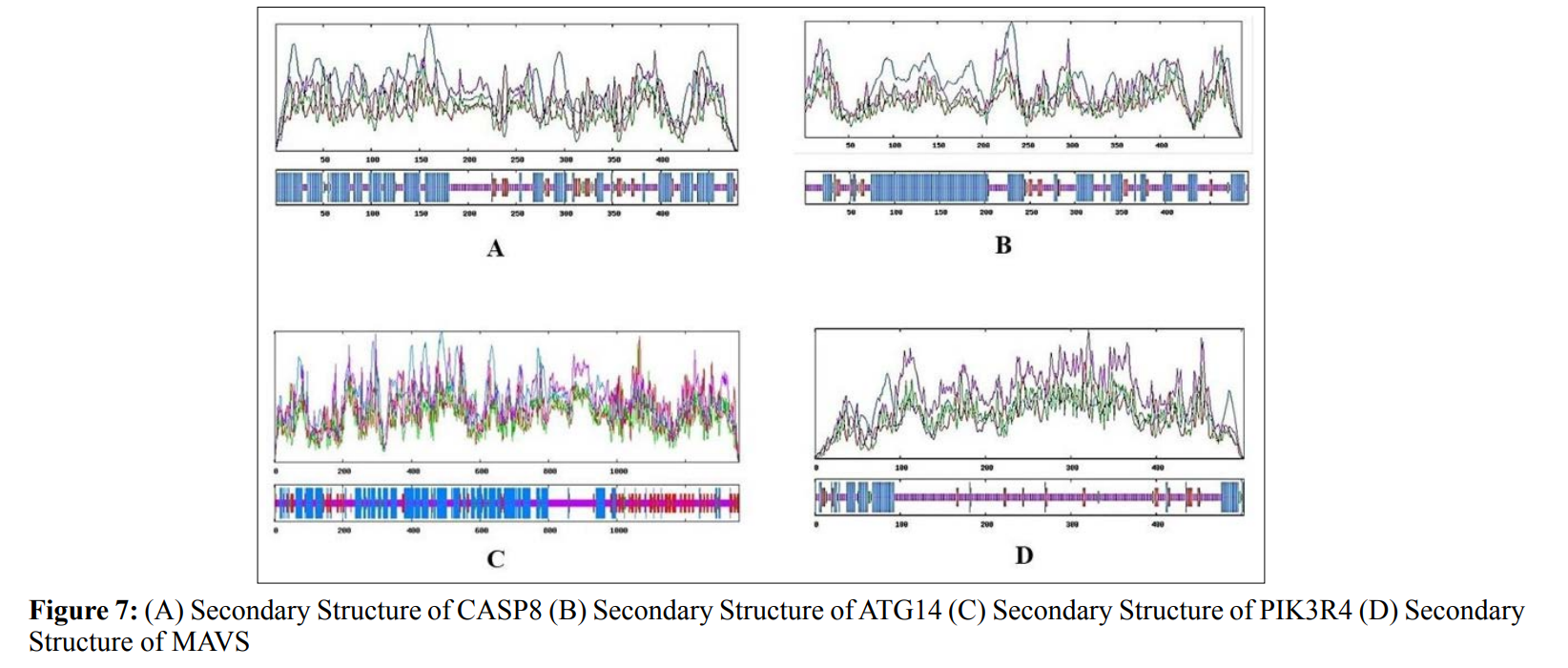
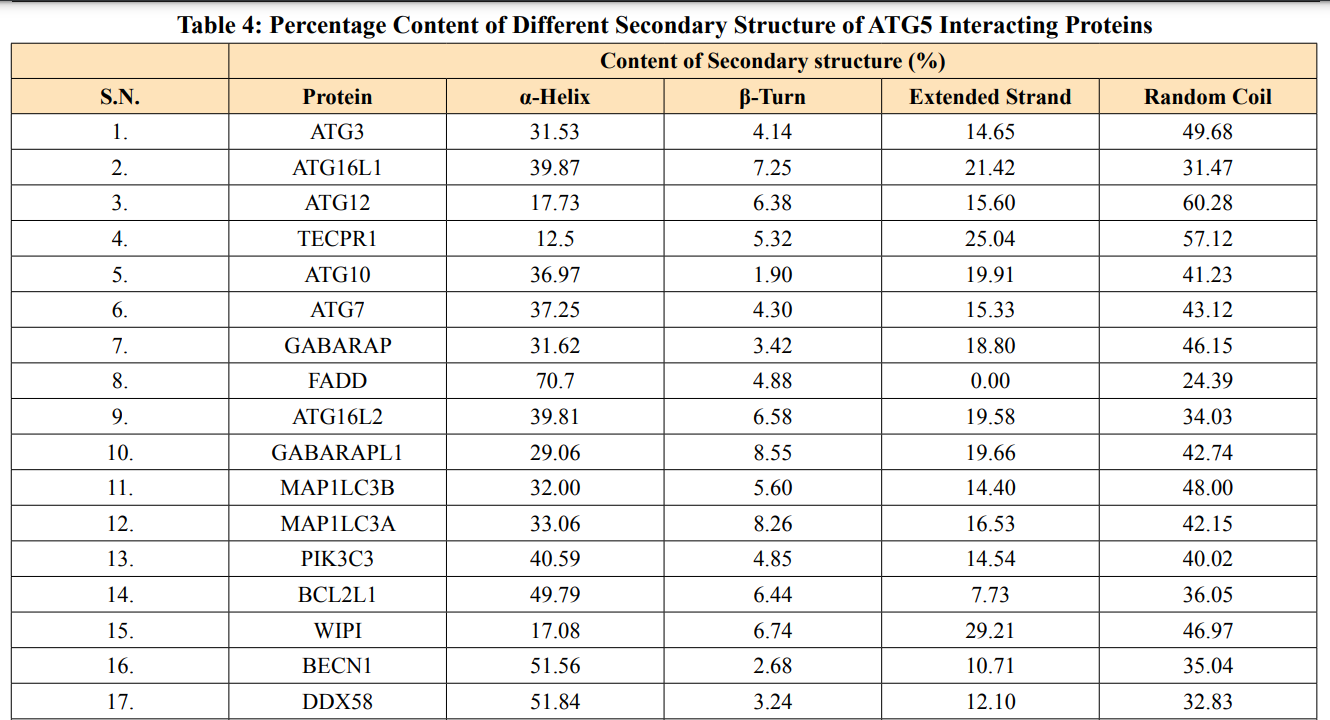
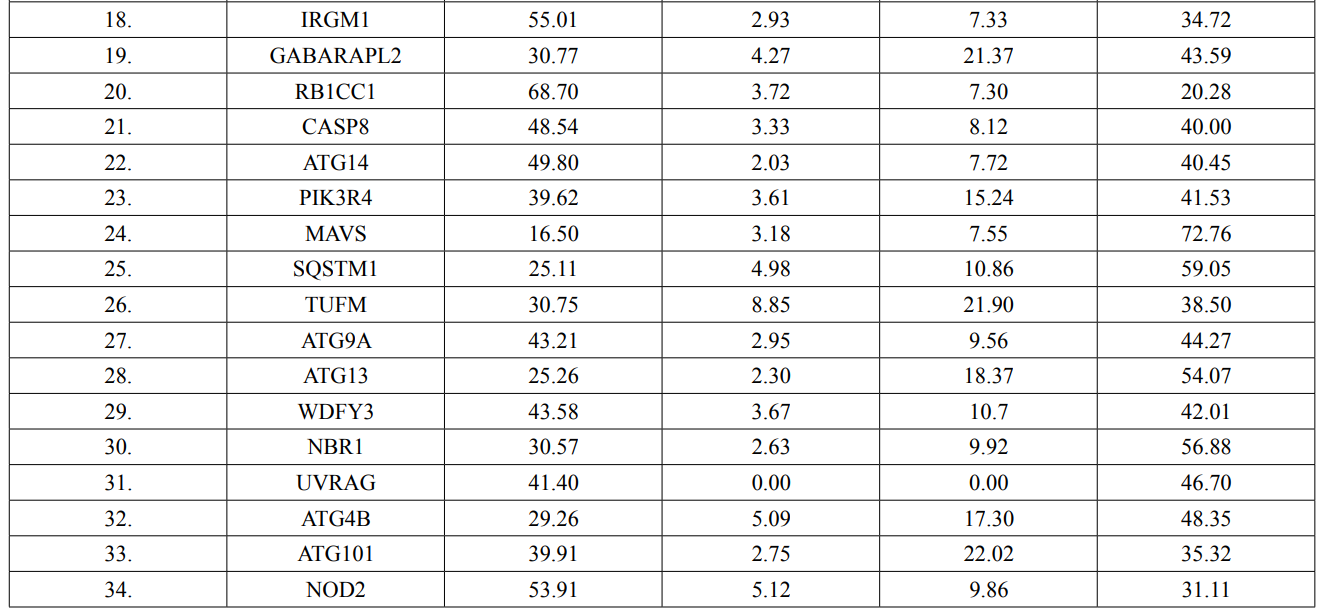
In this study, amino acids cysteine (C), tryptophan (W), methionine (M) and histidine (H) was found to be low in numbers among all thirty-four interacting proteins. This shows that these proteins cannot be analyzed using UV spectral methods [18]. The polypeptide backbone of local conformation proteins is referred to as secondary structure forming regular structures like α-helix, β-strand, random coils and extended strands. It is a critical step in predicting tertiary structure and gives crucial information about protein action [19]. ATG5 interacting proteins secondary structure predicted alpha helix and random coils were far more abundant than other secondary structures such as β-turn (Table 4, Figure 2-10).
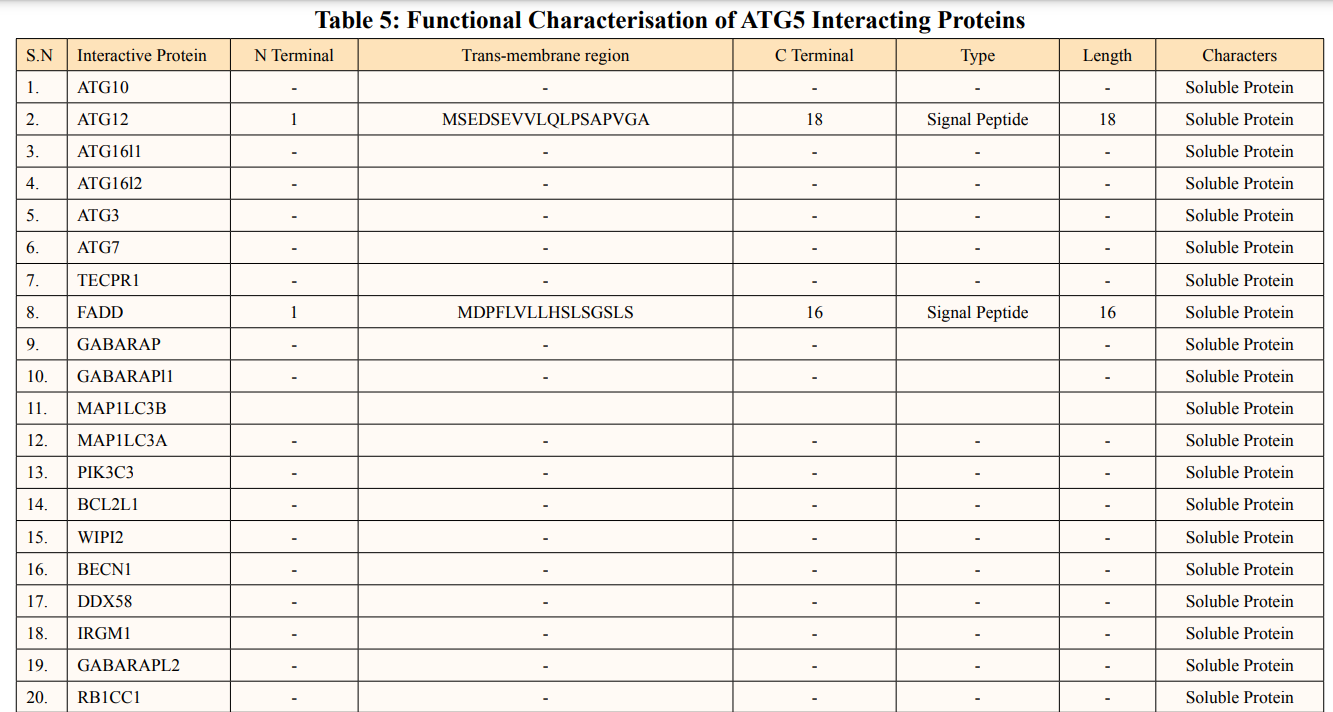
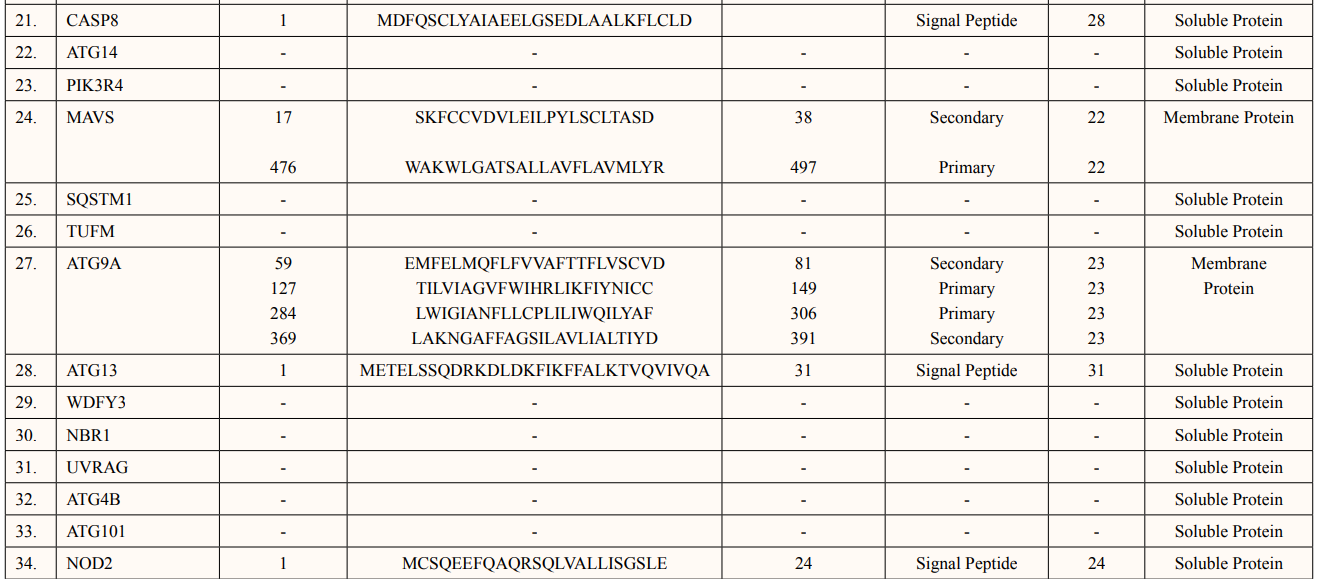
SOSUI server predicted functional characterisation of ATG5 interacting proteins (Table 5). ATG5 interacting proteins namely; ATG10, ATG16L1, ATG16L2, ATG3, ATG7, TECPR1, GABARAP, GABARAPL1, MAP1LC3B, MAP1LC3A, PIK3C3, BCL2L1, WIPI2, BECN1, DDX58, IRGM1, GABARAPL2, RB1CC1, ATG14, PIK3R4, SQSTM1, TUFM, WDFY3,NBR1, UVRAG, ATG4B and ATG101 were predicted as soluble protein. Moreover, ATG12 contains 1 Trans-membrane of length-18aa., FADD contains 1 Trans-membrane region of length-16aa., CASP8 contains 1 Trans-membrane region of length-28aa and ATG13 contains 1 Trans-membrane region of lengh-31aa, NOD2 showed 1 Trans-membrane region of length- 24aa. were predicted as signal peptides. There were two membrane proteins predicted of primary and secondary character namely: MAVS contains two transmembrane helices of length 23aa each and ATG9A having four transmembrane helices of length 23aa individually.
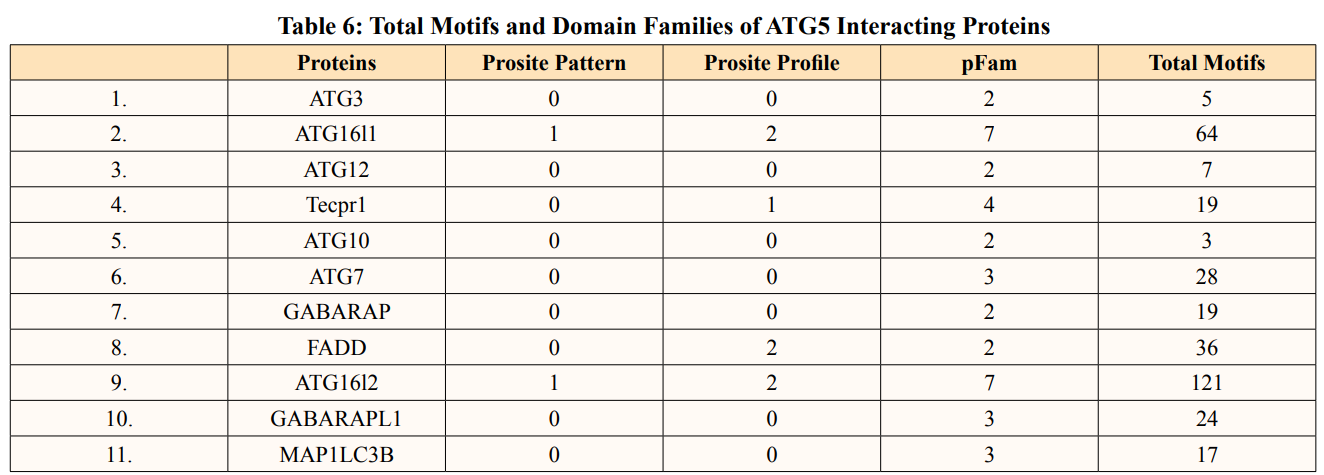
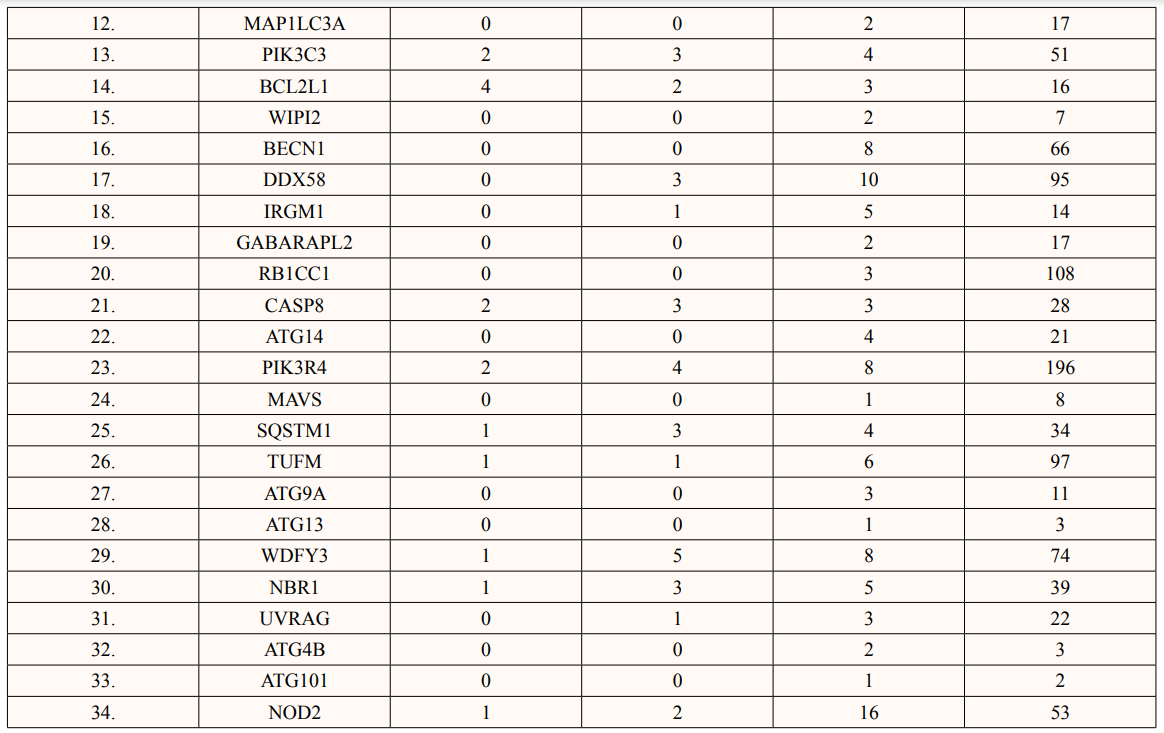
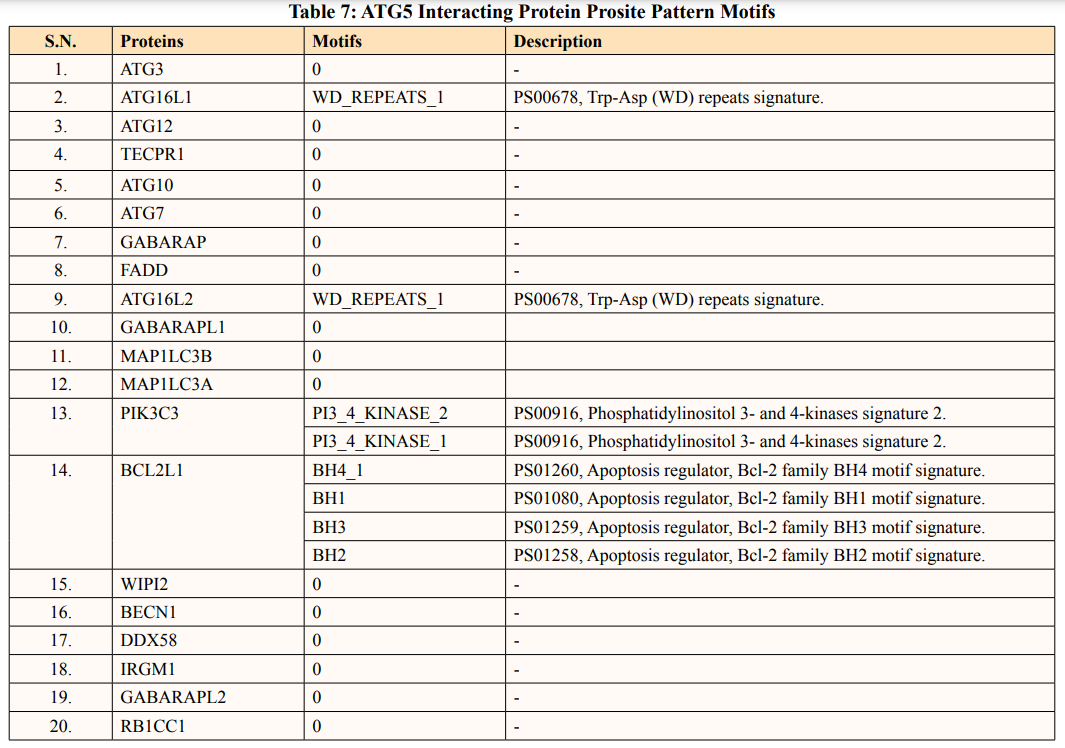
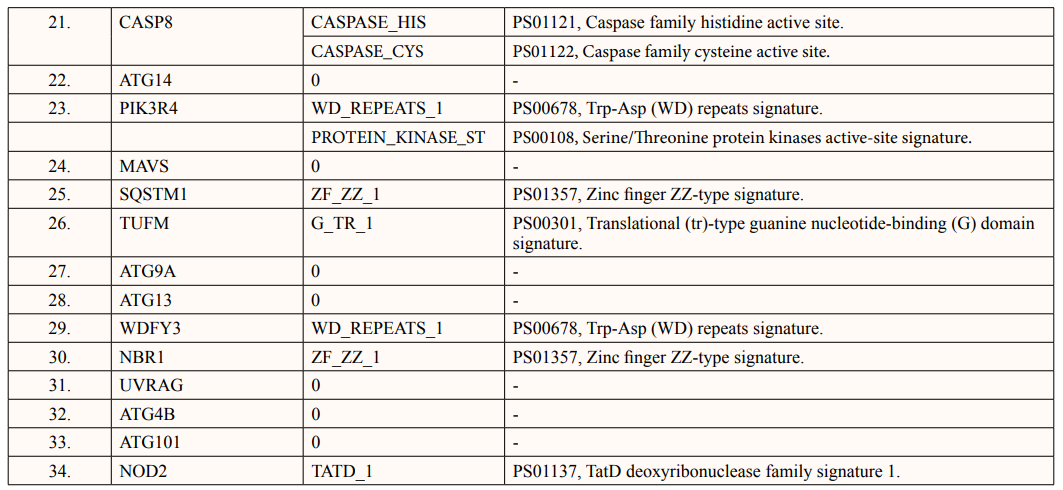
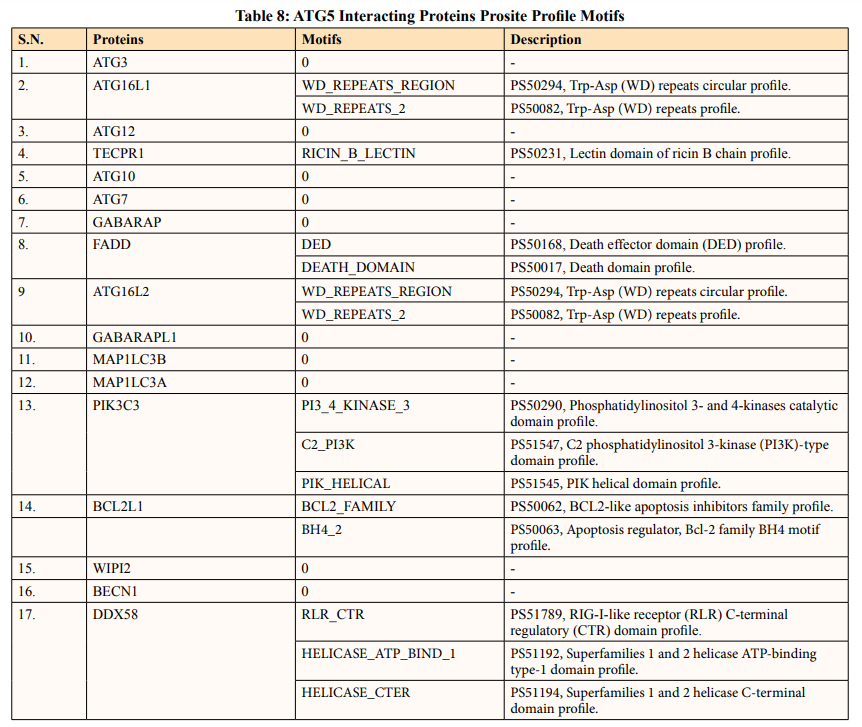
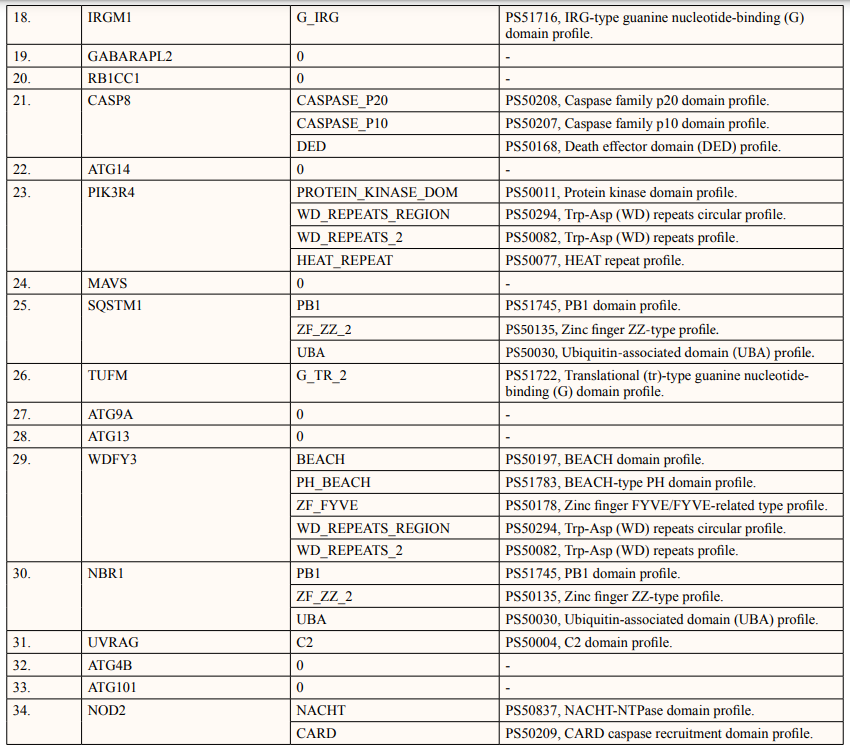
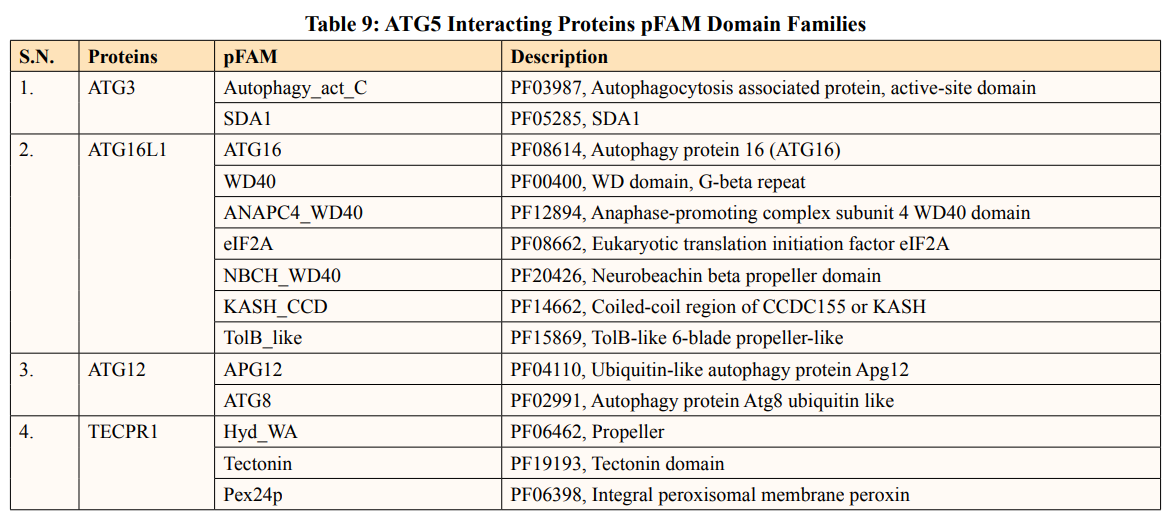
In this study, out of thirty-four ATG5 interacting proteins, ten proteins namely; ATG16l2, PIK3C3, BCL2L1, CASP8, PIK3R4, SQSTM1, TUFM, WDFY3, NBR1 and NOD2 showed both Prosite Pattern and Prosite Profile. The maximum number of Prosite Pattern and Prosite Profile was found in BCL2L1 and WDFY3 respectively. Further, 3 proteins viz, Tecpr1, FADD, DDX58 showed only Prosite Profile. However, there were seventeen proteins namely; ATG3, ATG10, ATG7, GABARAP, GABARAPL1, MAP1LC3B, MAP1LC3A, WIPI2, BECN1, GABARAPL2, RB1CC1, ATG14, MAVS, ATG9A, ATG13, ATG4B and ATG101 showed no Prosite Pattern and Prosite Profile. pFAM shows accurate group of protein domains and families [20]. The current study showed NOD2 an ATG5 interacting with sixteen protein families which have highest among all (Table 6-9).
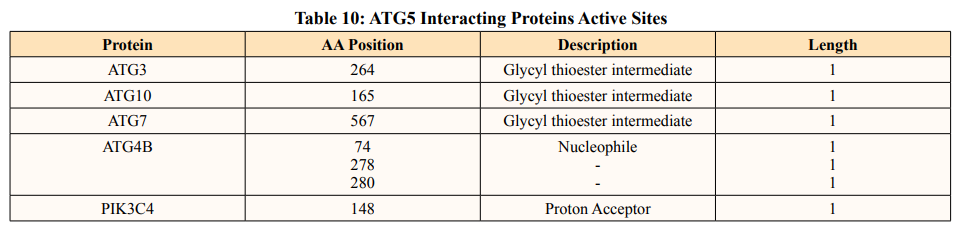
After binding of substrate to an active site of an enzyme, catalysis or chemical reactions take place. When a protein undergoes an enzyme reaction, active sites act as a structural feature that determines functioning of protein. In this study, total of five ATG5 interacting proteins i.e., ATG3(Position of aa-264), ATG10(Position of aa-165), ATG4B (Position of aa-74,278,280), ATG7(Position of aa-567) and PIK3C4(Position-148) showed active sites of length one amino acid independently (Table 10). ATG3, ATG10, ATG7 actively binds glycyl thioester intermediate whereas ATG4B showed an active site for nucleophile binding whereas PIK3C4 for proton binding.
In eukaryotes, autophagy is one of the main processes for proteins turnover. Few modulations in autophagy process may show diverse molecular functions of proteins. There is not enough full annotation for the genes and proteins involved in autophagy. Numerous genes implicated in autophagy might have different activities or be involved in different cellular processes. It is difficult to find appropriate biomarkers for autophagy and associated processes. Further, to track autophagic activity in cells and tissues, biomarkers are crucial. The assessment of autophagy in various experimental contexts is complicated by the absence of well recognised and validated indicators. However, these components attain a net change in cellular character. Therefore, it was critical to evaluate a protein’s interactions with other proteins while investigating their function. ATG5 have been interacted with ATG1 and ATG18 which reverses oxidative stress triggered synapse overgrowth [21]. Nevertheless, along performing crucial protective function in many diseases, uncontrolled autophagy can lead to excessive degradation of cellular components causing cell death. ATG5 plays vital role for cell death function by interacting with FADD [22]. The complexities of neurological processes may be reduced by bioinformatics techniques. It is a continuous challenge to develop more complicated models that take into account the complex nature of neural circuits and connections. In numerous neurodegenerative disorders like Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, aggregation of misfolded proteins remains as marker. This accumulation directly disturbs basal neuronal autophagy level [23]. Further, ATG5 interacting protein ATG7 is an essential effector enzyme for autophagy function loss of which exhibit complex neurodevelopmental disorders like ataxia and developmental delay [24]. Taylor et al. reported that absence of ATG7 in muscles cells of human resulted in impaired LC3 lipidation and autophagy flux [25]. Demonstrated roles of ATG16l1 in neurogenesis and stem cell development which is one of the ATG5 interacting protein predicted in this study. Moreover, ATG5 interacting membrane protein ATG9A interacted with lipid droplets and involve in cell survival, mitochondrial maintenance and recovery of neurogenesis defects due to impaired autophagy. Depletion of ATG9 resulted in abnormal development of axon tracts in brain regions of mouse causing axon specific lesions [26].
The STRING database showed detailed study and integration of protein-protein interactions. The data shown in the prediction of amino acids number where, Leu (L) and Ser (S) were found highest in interacting proteins and Cys (C), His (H), Met (M) and Trp (W) were found low in count (Table 3). Low levels of essential amino acids which are the precursors of various neurotransmitters may show deprived signal transduction in mice [27]. Disulfide covalent bridges formed by cysteine-cysteine residues have an important role in folding along with stability of proteins. This results from an oxidative folding method that takes place in thiol groups cysteines. Studies have suggested ways to make proteins more stable by mutating cysteine. Moreover, the stability decreases when the natural disulfide links are broken [28]. Hence, majority of ATG5 interacting proteins were unstable in nature as they lack cysteine-cysteine disulfide bridges. Leucine is a strong activator of the mechanistic target of rapamycin (mTOR) pathway that promotes anabolic cellular processes and growth which further ameliorate protein catabolism by autophagy mechanism [29]. Leucine also contributes to protein thermostability nonetheless, serine is investigated to be predominantly synthesized in astrocytes and not in neurons, indicating that it is an essential amino acid for neurons [30]. ATG5 interacting protein negative charges (-R) were calculated using the aspartic and glutamic acid content, while their positive charges (+R) were calculated using the arginine and lysine content by ExPASY ProtParam tool. According to Table 2, the majority of ATG interacting proteins showed pI values lower than 7, predicting aspartic and glutamic acids constitutes ATG5 interacting sequence than arginine and lysine. The ATG5 interacting proteins’ aliphatic index ranged from 60.50 to 103.90. Therefore, proteins can maintain their stability in higher temperature range due to presence of high aliphatic index.
The secondary structures of proteins such as α-helical structure is composed of methionine(M), alanine (A), leucine (L), glutamate (E), and lysine (K) amino acids, whereas the electrostatic repulsion and steric hinderance between bulky side chains of isoleucine or charged side chains of glutamic and aspartic acid causes a protein to adopt a random coil conformation. The random coil is typically described as a folded chain area that is more flexible and dynamic than other secondary conformational structures [31]. β-strand are composed of tryptophan (W), tyrosine (Y), phenylalanine (F), valine (V), isoleucine (I) and threonine (T), furthermore, glycine (G) and proline (P) amino acids help to build the relevant turns [32]. The predicted findings suggest that the amino acids concentration is simply responsible for making the particular secondary structure of proteins. The percentage score of amino acid distribution of ATG5 interacting proteins concludes that α-helix and random coils were dominant over other secondary structures followed by β-strand and turns (Table 4) which supports the amino acid compositions.
Protein solubility is an important feature. Intrinsic parameters like amino acid sequence and non-intrinsic parameters like temperature, ionic strength and pH influence protein solubility [33]. Protein accumulation and folding in the cellular environment are incompatible with one another because overexpression of unfolded proteins causes inclusion bodies to form [34]. In addition, the given data suggested soluble signal peptides interacts with ATG5. These signal peptides attach with the N-terminus of desired protein that directs them to the periplasmic space and resolves improper folding and overexpression of protein [35]. Most of the ATG5 interacting proteins were hydrophobic in nature. There are various factors like hydrophobicity, protein molecules volume, electric charge responsible for protein stability. Hydrophobicity is one of the most important factors of transmembrane helices also ATG9A is a sole multispanning membranous protein responsible for the early phagophore expansion. It functions in the mobilization of lipid droplets to autophagosome and mitochondria, blocking of which leads to the inhibition of mitochondrial respiration and lipotoxicity [36]. The two transmembrane helices of the mitochondrial antiviral signaling (MAVS) protein have a significant function in antiviral and autoimmune responses. By directly attaching LIR motif to LC3, MAVS inhibits autophagy [37].
Conserved domains and families serve as a building block and can be recombined in different arrangements to make proteins with different functions, and often correspond to the 3D domains of a protein structure [38]. Protein domains and motifs can be computed using the prosite database. Prosite profile aims to characterize a family of protein or domain along with whole length as contrast to prosite pattern, which is restricted to a small region with strong sequence similarity [39]. Further, ATG5 interacting protein motifs, domains and families were assembled with respect to their common functionality. By closely observing the signature pattern of particular family it can be reliable to predict new proteins.
Protein-protein interaction (PPI) networks are used to locate the multiple molecular pathways and mechanisms that causes disorders. The closest interacting protein had the shortest node score of 0.999 namely: ATG16l1. It functions by interacting with ATG12-ATG5 to mediate the conjugation of phosphatidylethanolamine (PE) and activates LC3II. Protein structures are analyzed and their active sites are considered as the starting point for multiple PPI studies. In current study two interacting proteins namely: ATG4B and ATG9A showed catalytic activity. In addition, ATG5 interacting proteins like ATG3, ATG10 and ATG7 had active sites acting as glycyl thioester intermediate and ATG4B, PIK3C4 as nucleophile and proton acceptor respectively. PIK3R4 involved in initiation and maturation of autophagosomes and endocytosis predicted to have ATP binding site. The C-terminal lipidation of MAP1LC3 is mediated by ATG7, ATG3, ATG5 and ATG16L1, and their deletion reduces autophagy at the later or maturation stage of autophagosome production, resulting in a shortage of amino acids as well as rapid cell death within 24 h The conjugation of ATG3 occurs to LC3 to a lipid molecule, phosphatidylethanolamine (PE) on autophagosome membranes [40-46].
In the present study, ATG5 interacting proteins as represented in (Table 1), physicochemical and functional parameters were predicted using different computational tools. This study concludes that ATG5 interacting proteins were stable in higher temperature. Further, ATG5 interacting proteins were predicted to have higher percentage of α-helix and random coils. Furthermore, these proteins were predicted to be hydrophobic in nature. In addition, wet lab experiments are needed to confirm the binding of ATG5 with the in silico identified interacting proteins. This study provides limited information of the molecular mechanisms that control autophagy in higher eukaryotic cells. However, this study may be helpful to get a theoretical idea in developing certain biomolecules for autophagy. In conclusion, predicting different properties and binding sites may contribute a better understanding of ATG5 regulatory functions that may give potential target for docking studies.
1.Kim JH, Hong SB, Lee JK, Han S, Roh KH, et al. (2015) Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 11: 75-87.
2.Vellai T (2009) Autophagy genes and ageing. Cell Death Differ 16: 94-102.
3.Vij A, Randhawa R, Parkash J, Changotra H (2016) Investigating regulatory signatures of human autophagy related gene 5 (ATG5) through functional in silico analysis. Meta gene 9: 237-248.
4.Tang M, Liu T, Jiang P, Dang R (2021) the interaction between autophagy and neuroinflammation in major depressive disorder: From pathophysiology to therapeutic implications. Pharmacol Res 168: 105586.
5.Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M (2007) Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 3: 591-596.
6.Rao VS, Srinivas K, Sujini GN, Kumar GN (2014) Protein- protein interaction detection: methods and analysis. Int J Proteomics 2014: 147648.
7.Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, et al. (2014) Autophagic clearance of mitochondria in the kidney copes with metabolic acidosis. J Am Soc Nephrol 25: 2254-2266.
8.Collier JJ, Oláhová M, McWilliams TG, Taylor RW (2021) ATG7 safeguards human neural integrity. Autophagy 17: 2651-2653.
9.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, et al. (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032-1036.
10.Brandi V, Di Lella V, Marino M, Ascenzi P, Polticelli F (2018) A comprehensive in silico analysis of huntingtin and its interactome. J Biomol Struct Dyn 36: 3155-3171.
11.Paramanik V, Krishnan H, Thakur MK (2018) Estrogen receptor α- and β-interacting proteins contain consensus secondary structures: An in-silico study. Ann Neurosci 25: 1-10.
12.Kurrey K, Paramanik V (2018) Identification and physiochemical analysis of ERK interacting proteins using bio-computational tools. World J Neurosci 8: 303-313.
13.Ma Y, Liu Y, Cheng J (2018) Protein secondary structure prediction based on data partition and semi-random subspace method. Sci Rep 8: 9856.
14.Hilario EC, Stern A, Wang CH, Vargas YW, Morgan CJ, et al. (2017) An improved method of predicting extinction coefficients for the determination of protein concentration. PDA J Pharm Sci Technol 71: 127-135.
15.Hoda A, Tafaj M, Sallaku E (2021) In silico structural, functional and phylogenetic analyses of cellulase from ruminococcus albus. J Genet Eng Biotechnol 19: 58.
16.Chang KY, Yang JR (2013) Analysis and prediction of highly effective antiviral peptides based on random forests. PloS one 8: 70166.
17.Zhang Z, Chow SY, De Guzman R, Joh NH, Joubert MK, et al. (2022) A mass spectrometric characterization of light- induced modifications in therapeutic Proteins. J Pharm Sci 111: 1556-1564.
18.Roy S, Maheshwari N, Chauhan R, Sen NK, Sharma A (2011) Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation 6: 315-319.
19.Hulo N, Bairoch A, Bulliard V, Cerutti L, De Castro E, et al. (2006) The PROSITE database. Nucleic Acids Res 34: 227-230.
20.Milton VJ, Jarrett HE, Gowers K, Chalak S, Briggs L, et al. (2011) Oxidative stress induces overgrowth of the Drosophila neuromuscular junction. Proc Natl Acad Sci USA 108: 17521- 17526.
21.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, et al. (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280: 20722-20729.
22.Collier JJ, Suomi F, Oláhová M, McWilliams TG, Taylor RW (2021) Emerging roles of ATG7 in human health and disease. EMBO Mol Med 13: 14824.
23.Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47: 147.
24.Wu X, Fleming A, Ricketts T, Pavel M, Virgin H, et al. (2016) Autophagy regulates notch degradation and modulates stem cell development and neurogenesis. Nat Commun 7: 10533.
25.Yamaguchi J, Suzuki C, Nanao T, Kakuta S, Ozawa K, et al. (2018) Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 14: 764- 777.
26.Sarafeddinov A, Arif A, Peters A, Fuchsbauer HL (2011) A novel transglutaminase substrate from Streptomyces mobaraensis inhibiting papain-like cysteine proteases. J Microbiol Biotechnol 21: 617-626.
27.Gao X, Dong X, Li X, Liu Z, Liu H (2020) Prediction of disulfide bond engineering sites using a machine learning method. Sci Rep 10: 10330.
28.De Bandt JP (2016) Leucine and mammalian target of rapamycin-dependent activation of muscle protein synthesis in aging. J Nutr 146: 2616S-2624S.
29.Santhoshkumar R, Yusuf A (2020) In silico structural modeling and analysis of physicochemical properties of curcumin synthase (CURS1, CURS2, and CURS3) proteins of Curcuma longa. J Genet Eng Biotechnol 18: 24.
30.Sanjaya RE, Putri KDA, Kurniati A, Rohman A, Puspaningsih NNT (2021) In silico characterization of the GH5-cellulase family from uncultured microorganisms: physicochemical and structural studies. J Genet Eng Biotechnol 19: 143.
31.Lopez MJ, Mohiuddin SS (2023) Biochemistry, essential amino acids. In StatPearls Treasure Island (FL) StatPearls.
32.Trevino SR, Scholtz JM, Pace CN (2007) Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J Mol Biol 366: 449-460.
33.Nogalska A, D’Agostino C, Engel WK, Cacciottolo M, Asada S, et al. (2015) Activation of the unfolded protein response in sporadic inclusion-body myositis but not in hereditary GNE inclusion-body myopathy. J Neuropathol Exp Neurol 74: 538-546.
34.Juibari AD, Ramezani S, Rezadoust MH (2019) Bioinformatics analysis of various signal peptides for periplasmic expression of parathyroid hormone in E.coli. J Med Life 12: 184-191.
35.Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CD, et al. (2021) The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun 12: 6750.
36.Cheng J, Liao Y, Xiao L, Wu R, Zhao S, et al. (2017) Autophagy regulates MAVS signaling activation in a phosphorylation-dependent manner in microglia. Cell Death Differ 24: 276-287.
37.Mooney C, Davey N, Martin AJ, Walsh I, Shields DC, et al. (2011) In silico protein motif discovery and structural analysis. Methods Mol Biol 760: 341-353.
38.Sigrist CJ, Cerutti L, Hulo N, Gattiker A, Falquet L, et al. (2002) PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform 3: 265-274.
39.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, et al. (2008) The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell 19: 4762-4775.
40.Tanida I, Ueno T, Kominami E (2004) LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36: 2503-2518.
41.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, et al. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456: 264-268.
42.Roy S, Maheshwari N, Chauhan R, Sen NK, Sharma A (2011) Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation 6: 315-319.
43.Raazia R, Nijil RN (2017) Method for predicting transmembrane helices in protein sequences, international journal of engineering research & technology (ijert) ncetet 5.
44.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, et al. (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280: 20722-20729.
45.Nivetha R, Meenakumari M, Bhuvaragavan S, Hilda K, Janarthanan S (2021) In silico analysis of carbohydrate- binding pockets in the lectin genes from various species of Canavalia. Comput Biol Chem 92: 107477.
46.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, et al. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425-434.
View PDF