Author(s): <p>Aravind Reddy Kuchkuntla</p>
Castleman Disease (CD) is a spectrum of heterogenous hematological diseases that share characteristic clinical, histopathological, and immunological features. We present a case of a 61-y female with history of non-Hodgkin Lymphoma and hypothyroidism presenting with fatigue, generalized weakness, nausea, and poor appetite. On admission, physical examination was unremarkable, and labs were notable for hyperkalemia, hyperuricemia and worsening renal function. Imaging showed lymphadenopathy in neck, mediastinum, and left axilla along with mediastinal and retroperitoneal lymphadenopathy. Initial work up of serum electrophoresis was suggestive of multiple myeloma however bone scan did not reveal any lytic lesions. As biopsy results were pending, patient developed worsening cytopenia’s and a repeat serum electrophoresis showed a M spike with IgG lambda against a polyclonal background. Bone marrow biopsy showed polytypic plasmacytosis that was negative for HHV 8 and lymph node biopsy also showed polytypic plasmacytosis. Further work confirmed the diagnosis of idiopathic multicentric Castleman disease and patient was treated with silutuximab and responded well.
Castleman Disease (CD) is a spectrum of heterogenous hematological diseases that share characteristic clinical, histopathological, and immunological features. The first case of CD was reported in 1954, and several revisions in its description were reported until Fajgenbaum et al published a set of guidelines for its diagnosis and management in 2017 [1]. Historically, based on number of involved lymph nodes and organ involvement, CD was classified into unicentric and multicentric disease. Unicentric CD presents as a solitary localized mass in the mediastinum or cervical region with no systemic constitutional symptoms. In such patients, surgical excision is therapeutic and recurrence rate is uncommon.
Multicentric CD was first reported in 1978 in a patient who had generalized lymphadenopathy, splenomegaly and had HV type along with plasma cell infiltrates in all involved lymph nodes [2]. This subset of CD involves a single or multiple groups of lymph nodes and these patients invariably present with constitutional symptoms such as fatigue, likely due to excessive interleukin (IL)-6 expression and release from B cell germinal centers [3]. Multicentric CD is further classified based on the severity of presentation and its association with Human herpesvirus-8 (HHV8) infection. The majority of cases published till mid-1990s were defined as HH8 associated MCD, in patients with a concurrent HIV and HHV-8 infection [4].
The second subset of patients with negative HIV and HHV-8 infection is known as idiopathic MCD. The most recent addition to the spectrum is a severe form of iMCD that is associated with TAFRO syndrome (thrombocytopenia, anasarca, renal dysfunction and organomegaly) [5]. Until guidelines were published in 2014, CD was classified based on lymph node histopathology into hyaline-vascular type (HV), plasma cell (PV) and a mixed type called hyaline-vascular plasma cell type (HV-PC) [6]. CD is now classified into different histopathological types ranging from lymph nodes with atrophic germinal centers with hypervascularization to hyperplastic germinal cells with polytypic plasmacytosis.
Annual incidence of all types of CD is estimated to be between 6500 to 7700 with approximately 1650 MCD cases [7,8]. The exact etiology of CD is still unknown and several mechanisms have been proposed and are being investigated to date. The most widely accepted mechanism involves viral IL-6 mediated vascular proliferation and human IL-6 generation in germinal centers of hyperplastic lymph nodes, which eventually lead to a state of hypercytokinemia causing systemic inflammation and organ dysfunction [9,10]. Other proposed mechanisms contributing to the cytokine storm include the role of immune deficiency, auto immune, chronic low grade inflammation, paraneoplastic and infectious etiologies such as EBV, toxoplasma and mycobacterium tuberculosis [11].
Given the non-specificity of presenting symptoms and the overlap of clinical pathology findings with auto-immune, lymphoproliferative, infectious and IgG4 disease, diagnosis of iMCD is difficult and is a diagnosis of exclusion after histopathological examination. Most of the literature on iMCD is generated from individual case reports and therefore there are inconsistencies in the approach to diagnosis and management of patients with iMCD. To address the knowledge gap, in 2015, an international working group of 34 experts from 8 countries on 5 continents established internationalconsensus diagnostic criteria for iMCD, based on 244 individual cases clinical cases and 88 tissue samples from patients who had a diagnosis of iMCD. Diagnosis is made if the patient has one major criterion and at least 2 of 11 minor criteria, with at least 1 laboratory abnormality and exclusion of infectious, malignant, and autoimmune disorders that can mimic iMCD (such as acute Epstein-Barr virus, lymphoma, systemic lupus erythematosus).
Major criteria include multicentric lymphadenopathy and biopsyproven histopathology on the iMCD spectrum. Histopathological spectrum ranges from a constellation of hyperplastic or regressed germinal centers, often with widened mantle zones in an onion-skin appearance, prominent follicular dendritic cells (FDCs) occasionally appearing dysplastic, hypervascularization, or with polytypic plasmacytosis. All the cases must be HHV-8 negative by latency associated nuclear antigen. Minor criteria include elevated C reactive protein (CRP) or erythrocyte sedimentation rate (ESR), anemia, thrombocytopenia or thrombocytosis, hypoalbuminemia, renal dysfunction, or proteinuria, polyclonal hypergammaglobulinemia, constitutional symptoms, hepatosplenomegaly, effusions or edema, cherry hemangiomata or violaceous papules, and lymphocytic interstitial pneumonitis. Guidelines recommend against using IL-6 levels for diagnosis due to lack of sensitivity and specificity [12].
Here we aim to present a patient in whom the diagnosis of CD was delayed and was treated as lymphoproliferative disorder secondary to EBV infection for a few years prior to presenting to our facility. The diagnosis was made based on a guideline directed approach and patient was initiated on a most recently approved therapy for iMCD.
A 61-year-old female nursing home resident with past medical history of non-Hodgkin lymphoma and hypothyroidism presented to the emergency department for worsening fatigue and abnormal laboratory test results. Per outside records from an outside hospital was diagnosed with EBV-related lymphoproliferative disorder and cryoglobulinemia in 2014. The patient complained of generalized weakness accompanied with nausea, vomiting, and poor appetite that started 3 days prior to presentation. Physical examination was unremarkable. Laboratory tests on admission were significant for hyperkalemia (6.1mmol/L), hyponatremia (122mmol/L), azotemia (91 mg/dL), elevated creatinine (5.61mg/dL), hyperuricemia (23mg/dL), hyperphosphatemia (3 mg/dL). Complete blood count demonstrated a mild leukocytosis (12.3 x 10 9 /L), anemia (hemoglobin 8.1 mg/dL), and thrombocytopenia (platelet 129 x 10 9 /L). Liver function tests were within normal limits. On further chart review, we noted that the patient had CT soft tissue neck one month prior to admission which showed lymphadenopathy in the neck, mediastinum, and left axilla. CT chest showed mediastinal and upper abdominal lymphadenopathy, findings suggestive of a lymphoproliferative disorder. Hematology was consulted for evaluation of generalized lymphadenopathy.
Initially, the patient was treated for hyperkalemia and suspected tumor lysis syndrome, with concerns for relapse of B cell lymphoma. Initial work up included serum protein electrophoresis (SPEP) and immunofixation that showed lambda monoclonal protein M spike 4.3, gamma globulin gap with predominantly an elevated total IgG level at 4.3g/dL, and kappa to lambda light chain ratio of 0.8. Based on the initial electrophoresis pattern, the patient underwent investigations for multiple myeloma whilst lymph node and bone marrow biopsies were pursued for workup of generalized lymphadenopathy. Bone scan did not reveal any lytic lesions. The initial plan was to start chemotherapy for presumed diagnosis of multiple myeloma.
During hospitalization, she developed worsening cytopenia’s (leukocyte 2.9 x 109 /L, platelet x 109 /L). Coagulation markers and fibrinogen levels were within normal range. A repeat SPEP was done due to the equivocal finding of M spike. A repeat protein electrophoresis showed the following: Alpha 1.4 alpha 2.6, B2 0.6, gamma globulin 4, Albumin/globulin ratio 0.5, total protein 8.4, and albumin of 2.8. Per report, M spike could not be completely excluded, and immunofixation showed M spike with IgG lambda against a polyclonal background. Increased gamma globulin fraction is suggestive of lymphoma, plasmacytic syndromes, chronic inflammation, acute infections, or chronic liver disease.
Bone marrow biopsy showed hypercellular bone marrow and polytypic plasmacytosis (30 to 35% of bone marrow) which were HHV-8 negative occasional EBV early RNA (EBER)-positive. (Figure 1). Flow cytometry demonstrated polytypic plasma cells with a kappa to lambda light chain ratio 0.8, no aberrant CD56 expression of plasma cells, and T and B lymphocytes without significant immunophenotypic abnormalities or increase in blasts. Lymph node core biopsy showed polytypic plasmacytosis. (Figure 2 and Figure 3). Flow cytometry showed immature plasma cells/plasmacytoid lymphocyte population with polytypic light chain expression (Kappa to lambda ratio approximately 2.7). Immunostaining for IgG4 was only expressed in a small subset of plasma cells. This does not support the diagnosis of IgG4 related disease. Based on the presence of polytypic plasmacytosis, the diagnosis of MCD was considered. Further work up supported the diagnosis with elevated IL-6, ESR, hypoalbuminemia and negative work up for HHV8 and HIV, autoimmune disease, lymphoma, and POEMS syndrome. The patient therefore met 1 major criterion with 3 minor criteria to establish a diagnosis of multicentric Castleman disease. The patient was initiated on siltuximab 11 mg/kg over 1 hour every 3 weeks. After treatment, creatinine improved to 1.46 mg/dL, with normalized potassium (3.9 mmol/L), uric acid 3.4 mg/dL, LDH (164 U/L), and CRP (3.7).
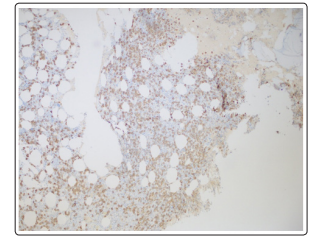
Figure 1: Increased plasma cells, CD 138 immunostaining on bone marrow biopsy
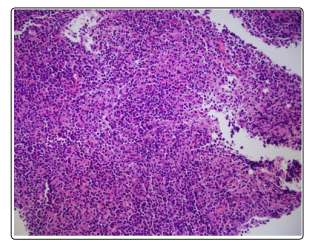
Figure: 2 Lymph node, increased plasma cells (H&E, x 200)
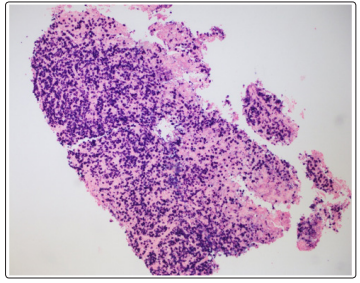
Figure 3: Plasma cells ISH kappa x 100, Lymph node biopsy
A recent systematic review on iMCD included data from 128 clinical case reports and data obtained from clinical trials (n=139). Common presenting features among these patients were fever (52%), lymphadenopathy (100%), organomegaly with hepatomegaly and splenomegaly (78%). Other less common presenting symptoms with 10-20% prevalence among patients were night sweats, weight loss and ascites with pleural effusions (74%-78%). Our patient presented with similar presenting features of fatigue, weight loss, and generalized lymphadenopathy with hepatosplenomegaly [13]. Biochemical abnormalities noted in the review were anemia (87% of patients), thrombocytopenia (44%), elevated ESR (92%), hypogammaglobinemia (77%), hypoalbuminemia (90%), elevated CRP (82%), and elevated IL-6 (90%). 80% patients also had elevated VEGF >100ng/L suggestive of abnormal vascular pathology. Our patient presented had all the laboratory abnormalities that have been most frequently reported with MCD.
In the same systematic review, the most common histology reported was plasmacytic subtype, accounting for 39% among case reports and 44% in patients in clinical trials, followed by mixed subtype (28% - 38%). A recent study from Castleman Disease Collaborative Network (CDCN) evaluated histopathology of 88 patients with a presumptive diagnosis of iMCD [12]. After extensive review, the CDCN has published a spectrum of histopathological findings that are graded 0 to 3. Five pathologic features are described: regression of germinal centers, prominence of follicular dendritic cell prominence, vascularity, and hyperplastic germinal centers and plasmacytosis. The regressed germinal center histopathology is consistent with HV type as described previously, whilst the follicular predominance with increased vascularity is classified as hyper vascular HV. Sheet-like plasmacytosis with hyperplastic germinal cells is identified as the plasmacytic subtype. Our patient presented with plasmacytic features in the lymph node biopsy.
As mentioned, CD is a diagnosis of exclusion. Work up should be negative for HIV serology, HHV8, EBER (lymph node), LANA -1 (lymph node), auto immune disorders, monoclonal gammopathy, and IgG4-related disease [12]. In our patient, work up was negative, however she was treated for EBV associated lymphoproliferative disorder 6 years prior to this presentation. We have reviewed the lymph node and bone marrow biopsy findings from the outside hospital; the findings were similar with plasmacytic predominance and polyclonal with few cells positive for EBV infection. The diagnosis of EBV related lymphoproliferative disease was made at the time shares a similar pathology as iMCD. We suspect that the diagnosed was missed as due to the lack of standardized guidelines for iMCD diagnosis at the time.
Management of patients with iMCD in the past was inconsistent due to lack of standard approach. Therefore, use of corticosteroids, chemotherapy with multiple regimens, radiotherapy and surgery have been used for treatment of iMCD. Current guidelines recommend treating the patient based on the severity of presentation [14]. Patients with 2 out 5 of the following features (Eastern Cooperative Oncology Group performance status >2, chronic kidney disease stage 4, anasarca, ascites, hemoglobin < 8mg/dL) or pulmonary involvement are considered to have severe iMCD. Patients with mild disease with who do not qualify for above criteria are classified as non-severe. Non-severe patients can be managed on an outpatient basis. Current recommendations advise the use of siltuximab, or tocilizumab based on the availability, as the first line agents for iMCD [15].
Rituximab with or without steroids can be considered in patients who failed first line treatment. For patients with severe disease, siltuximab or tocilizumab in combination with high dose steroids is recommended. Combination chemotherapy can be used in patients with poor performance status and requiring intensive care unit level care. Although these regimens have shown to have good response rates, they are associated with considerable toxicities and relapse rates. Treatment responses are evaluated based on CDCN criteria which include biochemical (decreased CRP, improvement of anemia and hypoalbuminemia), lymph node (regression on follow up imaging) and symptomatic resolution. Poor prognostic factors in these patients are age greater than 40 years, PC variant, and the presence of systemic signs such as anemia, pleural effusion, and hepatosplenomegaly [16].
Idiopathic multicentric Castleman disease is a rare entity of histopathological features, systemic symptoms, and biochemical abnormalities. iMCD shares numerous similarities to autoimmune and lymphoproliferative disorders and can easily be misdiagnosed. As in our case, iMCD can be mistaken for EBV-related lymphoproliferative disorder and can manifest in systemic symptoms as well as pancytopenia without adequate goal-directed therapy. We urge fellow clinicians to consider the diagnosis of iMCD even for patients with an established cause of generalized lymphadenopathy.
Conflict of Interest: None for all authors Patient consented for the publication of case report.