Author(s): Diana Khedr*, Sulaiman Alkhashan and Sharifa A.Alnaqeeb
Background: Focal seizures related to non-ketotic hyperglycemia (NKH) are rare in clinical practice. Plasma glucose levels are usually above 16.6 mmol/L and with normal or slightly elevated serum osmolality. The occurrence of focal seizures may be augmented by the absence of ketoacidosis. Electroencephalogram (EEG) during seizures usually confirms the diagnosis, however, the absence of epileptiform discharges does not rule out seizures. A non-ketotic hyperglycemiaassociated occipital lobe seizure can manifest itself as color flashes, blurry vision with periodic confusion, and usually resolves with insulin treatment and rehydration. Recently, seizures associated with nonketotic hyperglycemia have been found to be associated with subcortical T2 hypointensity on magnetic resonance imaging, especially in the occipital lobes. However, the mechanism remains unclear, although iron accumulation is suggested.
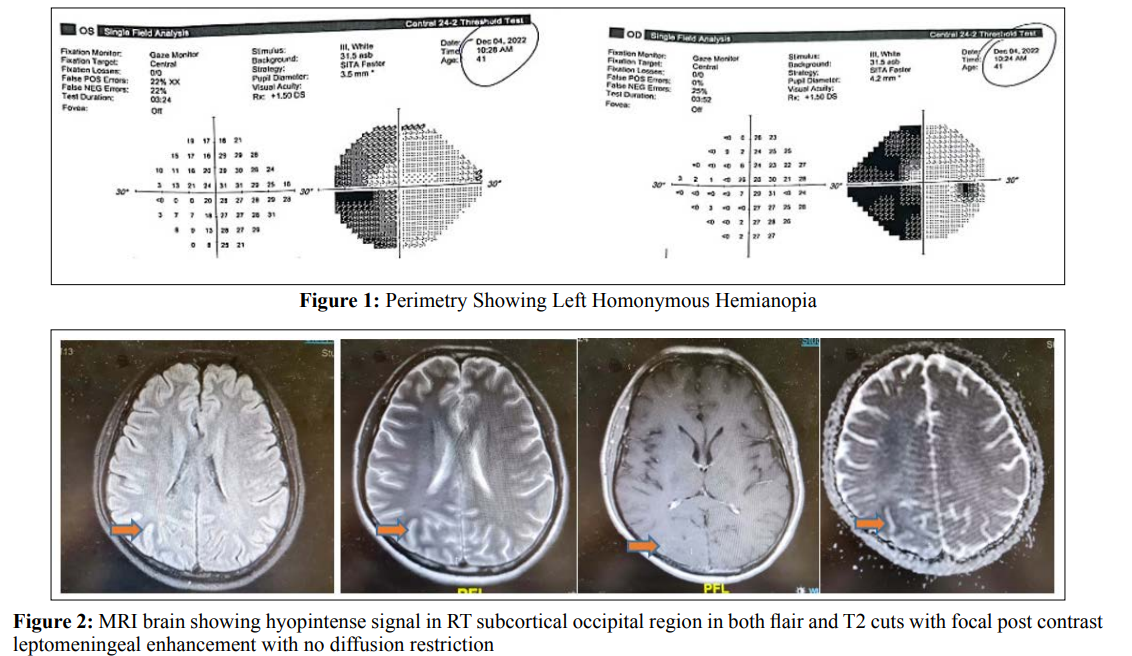
Case Presentation: We present a case 41- year-old male patient who presented headache, left-sided visual disturbances in the form of seeing round, colored flickering lights with left homonymous hemianopia and occipital lobe seizures associated with nonketotic hyperglycemia found to have a blood glucose of 18 mmol/L with a normal anion gap of 10 and calculated serum osmolality of 303 mOsm/L. Magnetic resonance imaging (MRI) brain showed subcortical T2 and flair hypointensity due to iron accumulation .The patient’s visual disturbances and seizures responded to rehydration and insulin treatment.
Conclusion: In conclusion, nonketotic hyperglycemia can be associated with occipital lobe seizures supporting the role of iron accumulation as a mechanism for subcortical T2 hypointensity in magnetic resonance imaging. Hyperglycemia should be taken into consideration when making an etiologic diagnosis of homonymous hemianopia.
Non-ketotic hyperglycemia (NKH)-related epileptic seizures can be diagnosed when high blood glucose is accompanied by normal plasma osmolality and negative urine ketone. The disease pathogenesis is not entirely clear. The possible mechanisms are hyperglycemia or hyperosmolality, a low level of gamma aminobutyric acid (GABA), and focal ischemia. Occipital lobe seizures have been described in the form of colored flashes or hallucinations, sometimes associated with aversive phenomena of the eyes and head to the side of the occipital lobe lesion. The neurological manifestations of severe hyperglycemia generally include choreoathetosis, ballismus, focal motor seizures and epilepsia partialis continua, coma [1-3]. Regarding vision loss, patients with diabetes often have visual abnormalities caused by diabetic retinopathy. Vision loss is generally classified into prechiasmal, chiasmal, or retrochiasmal deficits. The homonymous hemianopia observed in cases localized retrochiasmatically affects the right optic tract, lateral geniculate nucleus, optic radiations,or visual cortex. Hyperglycemic hemianopia has been rarely reported to cause temporary damage to the visual cortex, resulting in homonymous hemianopsia. Some reports have suggested an association of homonymous hemianopsia with epilepsy; however, the nature of the neurological impairment and neuroradiological findings in hyperglycemic hemianopia is not well known. Magnetic resonance imaging (MRI) classically demonstrates a low signal intensity along the subcortex on T2- weighted imaging. In previous studies, visual deficits improved or disappeared after controlling blood glucose levels in most of patients with homonymous hemianopia with hyperglycemia [4-7]. We herein report a case of irreversible homonymous hemianopia associated with severe hyperglycemia. We describe the disease process and discuss its underlying mechanism. Homonymous hemianopic visual field defects usually result from structural processes affecting retrochiasmal visual pathways. Cranial magnetic resonance imaging typically identifies the responsible lesions.
Written informed consent was obtained from the reported case A copy of the written consent is available for review by the Editor of this journal.
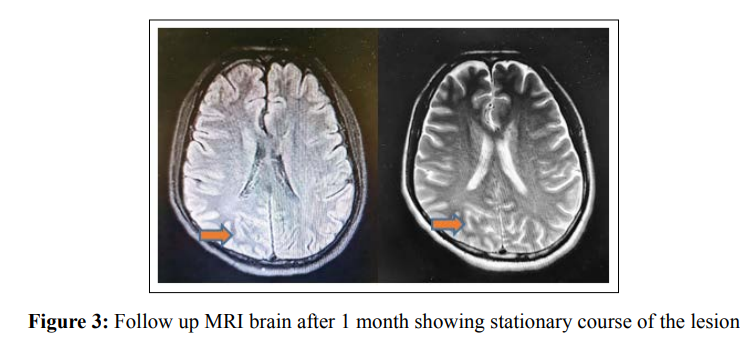
41 - year old male Indian patient, known diabetic for 10 years on oral hypoglycemic drugs and had past history 1 year before admission at our hospital of chest tuberculosis which was successfully treated with antituberculous medications for 6 months with total resolution of his symptoms, presented to our emergency department with subacute onset of severe headache, left-sided visual disturbances in the form of seeing round, colored flickering lights and once vomiting. His headache was mainly bi-frontal referred to whole cranium not associated with nausea, photophobia and phonophobia. On examination, He had left homonomous hemianopia with normal visual acuity and fundus examination, otherwise he had normal neurological examination. Initial computed tomography on brain, angiography and venography were unremarkable. Perimetry showed typical left sided homonomous hemianopia with macular sparing (Figure 1). He had a blood glucose of 18 mmol/L with a normal anion gap of 10 and calculated serum osmolality of 303 mOsm/L. His HBA1C was 14% with long standing uncontrolled non ketotic hyperglycemia. A lumbar puncture showed normal opening pressure of 18 cm H2O, normal cell count of less than 5 cell / mm3, normal proteins, sugar levels, negative TB PCR, negative culture and sensitivity and negative virology PCR. Two days after admission he was witnessed having generalized tonic clonic seizures with versive eye movement to the right which lasted less than one minute and was aborted. Brain magnetic resonance imaging (MRI) with IV gadolinium showed hypointense signal at T2 and Flair weight images involving right occipital subcortical white matter with mild leptomeningeal post contrast enhancement with no diffusion restriction (Figure 2). Interictal electroencephalogram (EEG) was unremarkable. Tight glycemic control by insulin and rehydration therapy were his main management plan, leviteracetam 500 mg twice daily dose was added for his provoked seizures as a short-term course till achieving good glycemic control. Follow up after one month from discharge, he had no more seizures with no more visual symptoms of flickering lights but still having left homonymous hemianopia. Follow up MRI brain showed stationary course regarding hypointense signal at right occipital subcortical region (Figure 3).
The onset of epileptic seizures in patients aged 40 years or more suggests a brain lesion as the initial hypothesis. One of the possible causes of seizures is a metabolic disorder such as nonketotic hyperglycemia in type 2 diabetes. Focal seizures induced by hyperglycemia were first reported in 1965 [8]. Nonketotic hyperglycemia can vary from asymptomatic to severely symptomatic, and its rapid recognition is vital, as treatment with insulin and aggressive rehydration can prevent serious outcomes [9, 10]. Diagnosis is also essential for the management of seizures because they are usually refractory to antiepileptic agents. In fact, some seizure medications, such as phenytoin, may even aggravate them because it interferes with insulin secretion causing hyperglycemic state. These seizures typically stop spontaneously after hyperglycemia is corrected [11]. Occipital lobe seizures have been described in the form of colored flashes or, more rarely, elaborate hallucinations sometimes associated with aversive phenomena of the eyes and head towards the side of occipital lobe lesion [12]. In addition, there are reports of aphasia associated with partial motor effects, pilomotor, and gyratory seizures [13]. Seizures associated with nonketotic hyperglycemia are often recurrent, and states of “petit mal” are seen in the form of epilepsia partial continua (EPC) [14, 15]. In these cases, the seizures tend to occur at an early stage of hyperglycemia while osmolality is still normal or only slightly elevated. Seizures usually stop once hyperglycemia is under control. The pathophysiology of epileptic seizures during nonketotic hyperglycemia remains unclear. The possible mechanisms are hyperglycemia or hyperosmolarity, a low level of gamma amino-butyric acid (GABA), and focal ischemia. Brick et al. suggested that the Krebs cycle in NKH is inhibited, resulting in increased GABA metabolism, which decreases GABA levels, thus lowering the seizure threshold causing neuronal hyperexcitability [16]. Another hypothesis involved the decrease of seizure threshold due to metabolic disturbances [17]. Hyperosmolality and dehydration induced by hyperglycemia or hypo-sodium accompanying hyperglycemia were suggested to trigger focal seizures and lead to a neurological deficit in some patients [18]. Hyperglycemia creates a hyperosmolar gradient between the intra and extracellular neuronal environments, thereby facilitating seizures. In addition to that, hyperglycemia increases GABA metabolism and thereby diminishes the seizure threshold [19-21]. ATP-sensitive potassium channels are also thought to be responsible for neuronal hyperexcitability and the precipitation of seizures in hyperglycemia [22]. Astrocytes cultured in a high-glucose environment show low mRNA expression of the Kir4.1 potassium channel, but a restoration of the normal glucose concentration normalizes the expression after a few days. In addition, glial glutamate uptake is low in a high glucose environment. Thus, low glutamate uptake in combination with poor potassium clearance, because of low Kir4.1 expression might lead to excitotoxic damage to neurons [23]. Previous studies have also suggested that disruption of the blood-brain barrier plays a role in the pathogenesis of the neurological signs described above. Gyral and leptomeningeal contrast enhancement has been observed, and may result from a disruption of the blood-brain barrier and extravasation of contrast medium, because of the greater metabolic activity during seizures. The hypoxic ischemic state that is caused by hyperglycaemia could lead to excitotoxic axonal damage and the accumulation of free radicals or iron, causing a hypointense signal on T2-weighted and flair images, which could also be caused by intracellular osmotic dehydration [24]. The presence of diffusion restriction in many cases of NKH suggests that cytotoxic oedema may be an underlying pathogenetic mechanism. Posterior leukoencephalopathy syndrome also affects the occipital lobe, which is attributed to autonomic dysfunction.
Sympathetic nerve innervation increases cerebral vascular tone in response to blood pressure increase. Less abundant sympathetic innervation of the posterior circulation would result in posterior predominance [25]. Patients with diabetes mellitus may be at risk of autoregulatory failure due to sympathetic dysautonomia and endothelial dysfunction [26]. The mechanism underlying the visual deficit in patients with severe hyperglycemia may be attributed to the disruption of autoregulation in the posterior circulation. Electroencephalography (EEG) in contrast to the generalised seizures that categorise hypoglycemia, hyperglycaemia in general causes focal seizures, which may be related to the presence of K-ATP channels in certain parts of neocortex [27- 29]. EEG generally reveals focal epileptiform activity with sharp or spike wave activity apparent on the posterior cortical leads. Unilateral or bilateral, asynchronous or synchronous focal epileptiform discharges are most common in the occipital region, followed by the temporal and parietal regions. Patients can also develop EPC which is often characterised by continuous focal epileptic activity on EEG, and generalised or focal slowing is common [30, 31]. However, patients can also display normal electroencephalograms as seen in our patient. The absence of ketosis is also essential; seizures in patients with ketoacidosis are very rare. The acidosis increases the bioavailability of GABA by increasing the activity of the enzyme that syntheses it and by decreasing its transamination [32]. GABA bioavailability is reduced in the absence of ketoacidosis and, therefore, the seizure threshold is minimized. This role of acidosis is useful in the treatment of some pediatric partial seizures, and these patients are prescribed a ketogenic diet [33]. Using antiepileptic medications is generally not justified. Moreover, cases of EPC are typically unresponsive to antiepileptic medications. Meanwhile, the rapid correction of hyperglycemia stops seizures very effectively [34].
A diagnosis of cortical lesions secondary to NKH may be delayed because of a lack of awareness of the various manifestations, and may lead to inadequate management and a lack of full recovery. Therefore, it is important to perform a detailed clinical evaluation and brain MRI in patients with an abnormal blood glucose concentration and visual or other cortical symptoms. It is possible that many of the cases reported previously might have had some irreversible cortical vision loss or field defects. In summary, the symptoms of NKH are referable to the posterior cerebral cortex. Various types of focal seizures can be seen clinically, and occipital seizures and various visual defects are the most common symptoms, with irreversible vision loss being possible. The disease is characterized by specific MRI findings that are diagnostic for this metabolic derangement.
Conflict of Interest: On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding Sources: No fund was needed for this study
Author contribution: DK, SA and S Alkhashan responsible for data collection, analysis, interpretation of results and drafted the article. S Alkhashan revised and approved the version to be published.