Author(s): Abdullah Al-Zahrani*,Mohammad Abdullah Alhasani
A 23 short-stature female patient presenting initially to gynecologist for left ovarian mass and bilateral sciatica but on further investigations it had been proved to be big pelvic neurofibroma with little findings of neurofibromatosis type 1. Surgical excision had been done without any complications. We aim to discuss the manifestations of neurofibromatosis type 1 and why lack of some findings in the current case with the role of neuroimaging in diagnosis of the case.
VARIOUS types of neurogenic tumors can affect the abdomen and pelvis. These tumors can be classified as being of ganglion cell origin (gangli- oneuromas, ganglioneuroblast-omas, neuroblasto- mas), paraganglionic system origin (pheochromo- cytomas, paragangliomas), or nerve sheath origin (neurilemmomas, neurofibromas, neurofibromato- sis, malignant nerve sheath tumors) [1-3]. These neurogenic tumors usually follow the distribution of the sympathetic ganglia along paraspinal areas or arise from the adrenal medulla or the organ of Zuckerkandl. Occasionally, however, other sites (eg, urinary bladder, bowel wall, abdominal wall, gallbladder) can be involved [3]. The urinary bladder is an unusual site of involvement because the trigone of the bladder contains cells of neural crest origin.
NF 1 is the most frequently-observed phakoma- tosis, with a frequency of occurrence of approxi- mately 1 in 3300. Half of the cases occur via autosomal dominant inheritance, and the rest occur as a result of spontaneous mutations. The NF 1 gene is located on chromosome 17 and encodes a protein called neurofibromin, which functions as a tumor suppressor [4-6]. Tumoral formations ob- served in NF1 occur due to mutations in this gene [1,3]. The diagnostic criteria were established at the National Institutes of Health (NIH) Consensus Development Conference in 1987 (Table 1) [7].
1. The presence of six or more cafe au lait macules 5mm or more in size during prepuberty, or 15mm or more in size during postpuberty.
2. The presence of two or more neurofibromas of any type, or the presence of one plexiform neurofibroma.
3. The presence of freckling in the axillary and inguinal regions. 4. The presence of optic glioma.
5. The presence of two or more Lisch nodules (iris hamartoma).
6. The presence of bone anomalies, such as long bones with thin cortex without arthrosis, together with sphenoid aplasia or pseudoarthrosis.
7. The presence of NF1 diagnosis in first-degree relatives according to the diagnosis criteria written above. Two or more of the above points should be present in the cases diagnosed with NF1. The current study presents the diagnosis, and treatment of paraspinal pelvic neurofibromas.
A 23 short-stature female patient complaining of dyspareunia, urgency in urination, and backache. She seeked medical advice at her gynecologist who examined her and proved to be free. Then, she was referred to orthopedic who suspected neuro- fibromatosis type I according to Table (1), especially skin manifestations seen in Figure (1), cutaneous neurofibromas [NFBs] of the limbs and trunk, and Freckling, or Crowe sign, in the inguinal region.
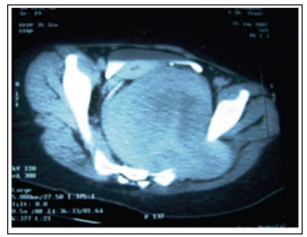
Ophthalmic examination revealed the Lisch nodule using slit lamp. She was subjected to radiological examination which revealed: Pelvic CT with contrast: Huge retroperitoneal oval-shaped solid lesion in straight contact with the anterior part of the sacrum (Figure 2) and displacing both uterus and rectum to the right. The fat plane was obliterated between the mass and the left sacral foramina, which was rela- tively enlarged and contained soft tissue isodense with the mass. Magnetic resonance imaging showed a 8x7x7.5 cm presacral tabulated mass as intermediate inten- sity on the T 1 -weighted images and high intensity on the T2-weighted images with a well-circums- cribed taw intensity center. The mass has expanded the left S-2 neural foramen into the spinal canal and encircled the S-2 neural root epidurally. The mass within spinal canal and sacral foramina was totally enhanced and the intrapelvic mass partially enhanced after contrast medium injection (Figure 3).
Figure 1: Presence of more than 6 café au lait bullae on the skin
>
Figure 2: Pelvic CT with contrast showed huge retroperitoneal
oval- shaped solid lesion in straight contact with the anterior part of the sacrum and displacing both uterus and rectum
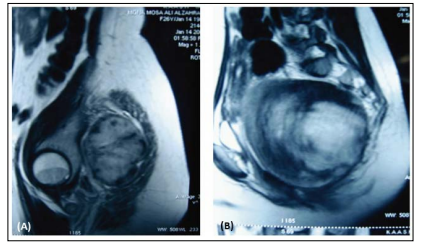
Figure 3: MRI images showing a huge pre- sacral lobulated mass as interme- diate intensity on the T1-weighted image (A), and high intensity on the T2-weighted image with a well-circumscribed low signal in- tensity center (B)
Posterior approach: Excision of S2 left side. Anterior approach: Dissecting the tumour and its weight was 1.8Kg and it reaches the great hiatus foramina. An extra-peritoneal approach through a right J incision obtained adequate exposure deep in the presacral space with the help of obstetricians.
A well-circumscribed giant mass emanating from the first neural foramen of the sacrum was found at the level of promontorium and displaced the uterus and rectum to the right (Fig. 4). The tumor originated from the S-2 root fibers. The mass was almost totally excised, except for the intrasacral portion, by sacrificing the intervening S-2 root fibers.
The well-circumscribed, oval-shaped firm mass was white yellow with homogeneously gray and gelatinous cut surfaces. Histological examination revealed moderate cellularity with fusiform and revealed moderate cellularity with fusiform and elongated cells containing many dark-stained nuclei embedded in a collagenous matrix. There were many thickened and hyalinized vessels, and a few inflammatory lymphocytes. Immuno-histochemi- stry showed strong vimentin and fibronectin posi- tivity, but no S-100 protein, cytokeratin, glial fibrillary acidic protein (GFAP), desmin, or factor VHI-related antigen staining, which confirmed the diagnosis of neuro-fibroma (Figure 4).
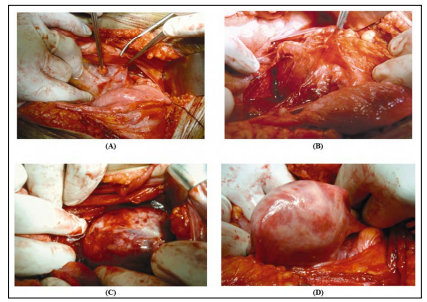
Figure 4: Operative excisional steps of the mass
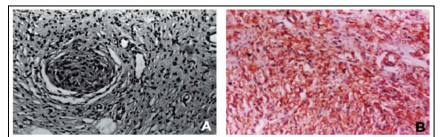
Figure 5: A- Photomicrograph of the neurofibroma consisting of fusiform and elongated cells with dark-stained nuclei embedded in collagenous matrix. Thickened hyalinized vessels and few inflammatory lymphocytes are also seen. IIE stain, x200. B- A vimentin-positive area is seen in the center, x200
The postoperative course was uneventful and the patient was discharged on the postoperative 15th day with no neurological deficit, but ongoing pain. Her complaints had resolved at the follow- up examination after 5 months and she was doing well. Postoperative MR imaging delineated a small in-trasacral residual tumor mass visible on both T2- and T 1 -weighted images.
The NF 1 gene is found on chromosome 17 and encodes a protein called neurofibromin, which is a tumor suppressor [1-4,8]. Mutations in this gene can result in decreases of this protein by different amounts, leading to development of different types of NF and to the various tumoral lesions seen in NF. Watson syndrome, NF-Noonan syndrome and segmental NF 1 are accepted as NF 1 variants [4,5, 8,9]. The mutation of the NF 1 gene results in either a loss of function or a nonfunctional neurofibromin protein that allows for Ras activation and ultimately for uncontrolled cell proliferation [10]. The defi- ciency of neurofibromin affects cell types through- out the entire body, especially the neural crest cells that include Schwann cells, melanocytes, and en- doneurial fibroblasts. Additionally, heterozygosity can be lost-a second-hit mutation to the normal NF1 allele-resulting in 2 abnormal NF1 genes [11]. According to several studies, this process seems to be the mechanism behind the formation of neu- rofibromas and cafe ?au-lait macules (CALMs) [12]. However, this mutation is not the cause of all of the features of this disease; current studies are still trying to elucidate the exact mechanisms. The actual timing of this acquired mutation is unknown.
The diagnostic criteria for neurofibromatosis- 1, developed by the National Institutes of Health (NIH) Consensus Conference in 1987, are based on clinical findings. The hallmark features of the disorder are CALMs and neurofibromas [1,7]. The NIH criteria are highly sensitive and specific in all age groups except in young children. DeBella et al., showed that 97% of children older than 8 met the diagnostic criteria, but children under 8 years old often do not; therefore, these criteria may need to be modified for children in this age group [13].
“Café au lait” spots are seen in 95% of patients, and axillary and inguinal freckling is observed in 70% of adult patients with NF1 [2,10]. Both types of spots were detected in the current case report. CALMs are hyperpigmented, flat, well-demarcated lesions of the skin that arise during the first year of life. These macules increase in both size and number with age [14]. The number of these lesions stabilizes over time, and the lesions tend to fade in older adults, similar to our patient. Current evidence shows the pathoetio-logy behind CALMs to be the presence of a mutation in both copies of the NF 1 gene in melanocytes, resulting in giant melanosomes and increased melanin [2]. Freckling, or Crowe sign, usually appears by 4 years of age and most commonly occurs in the axillary and inguinal regions but can occur in any intertriginous region [15]. The Lisch nodule, or melanocystic hamartoma in the iris, in the most frequently seen ophthalmo- logic feature in NF and is observed in 95% of cases which is the case in our study, but no optic glioma. These two eye conditions are among the important diagnostic criteria for NF [2]. The slit-lamp oph- thalmoscopic examination is the best method for finding these Lisch nodules [11].
Neurofibromas can grow focally or spread along the length of the nerve [1,9]. These tumors most commonly form in the skin but can grow anywhere in the peripheral nervous system and can involve any organ. Plexiform neurofibromas can cause complications such as pain, nerve root and spinal cord compression, and vertebral erosion [15,16]. Both subcutaneous and plexiform neuro-fibromas have the potential to transform into malignant peripheral nerve sheath tumors (MPNSTs) [2,18]. The overall risk of malignancy in neurofibromato- sis-1 patients is 2.5 to 4 times more than that of the general population, but this increased risk applies only to brain and connective tissue tumors [11,13]. Interestingly, the risk of cancer is higher in female neurofibromatosis-1 patients versus males [2,18,19].
The skeletal abnormalities often seen in neu- rofibromatosis-1 patients include pseudarthroses, bone lesions, scoliosis, osteoporosis, short stature, macro-cephaly, prominent brow and forehead, and pectus excavatum [2,4,6]. The short stature of our patient is consistent with the GH deficiency and early puberty related to NF. Neurofibromas in the retroperitoneal and pelvic regions may cause ureteric or urethral obstruction and result in hydronephrosis [2]. These tumors may pose difficulty for catheterization. Our patient seeked gynecologic advice at first.
MR imaging can be of great value in diagnosing such extensive neurogenic tumors when multiple ringlike structures are seen within the masses on T2-weighted images, likely representing nerve tissue and areas of myxoid degeneration, or markedly increased signal intensity with multiple hypointense septations as quite specific for plexi- form neurofibroma when observed in a diffusely infiltrating pelvic mass similar to the current case (target sign) [1-3, 16].
Complete resection is recommended to prevent the local recurrence and malignant transformation. Neuroradiological imaging is highly recommended for diagnosis and preoperative assessment.