Author(s): Esther Brigitte Ovaga*, B Mohammed Sidi, SI Harouna, PM Mulendele, M Njie, M Haboub, L Azzouzi and R Habbal
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is a cardiomyopathy of genetic origin, caused by abnormalities of desmosomes, characterized on the physiopathological level by a fibro-adipose infiltration replacing the myocardium of the right ventricle and the clinical level by an electrical instability leading to ventricular arrhythmias. ARVC/D peaks in frequency between the ages of 30 and 50. Diagnostic criteria have been established to retain the diagnosis of ARVC/D. Imaging and especially magnetic resonance imaging (MRI) play an important role in this diagnosis. We report the observation of a 48-year-old man, a former smoker, with a family history of the sudden death of a sister during a bicycle race, and who has been complaining for several years of palpitations. Clinical presentation, electrical signs, cardiac ultrasound and imaging findings lead to the diagnosis of ARVC/D. According to this observation, the authors describe the authors review the literature of this rare entity and discuss the different therapeutic approaches.
During the last decades of progress in the field of cardiology, many newly declared diseases have been described. One of these newly recognized conditions was arrhythmogenic right ventricular dysplasia (RAVD), which was first described [1]. The clinical spectrum of this condition has been described [2]. The typical prevalence is between 1/2000 and 1/5000 patients. DAVD peaks in frequency between the ages of 30 and 50. Despite its low prevalence, it is said that 5-20% of sudden deaths in young people are due to AVDD. Patients present with different symptoms, especially after puberty and before the age of 50. In addition, a family history is present in 30-70% of cases due to autosomal dominant inheritance.
DAVD is a cardiomyopathy of genetic origin, caused by abnormalities of the desmosomes, characterized on the physio pathological level by a fibro-adipose infiltration replacing the myocardium of the right ventricle and on the clinical level by an electrical instability leading to ventricular arrhythmias [3]. Diagnostic criteria have been established to retain the diagnosis of DAVD. Imaging and especially magnetic resonance imaging (MRI) play an important role in this diagnosis.
We report the observation of a 48-year-old man, a former smoker, with a family history of the sudden death of a sister at the age of 26 during a bicycle race, complaining for several years of palpitations.
These are angina-like palpitations of sudden onset and end, occurring with minimal effort as well as at rest, well tolerated functionally, associated with progressively worsening stage II dyspnea becoming stage IV with orthopnea. The patient does not report any notion of vertigo, syncope or faintness. The physical examination notes a normotensive patient (BP at 115/85 mm Hg) with a heart rate at 130 beats/min. Cardiac auscultation finds a systolic murmur rated 3/6th at the pulmonary focus. Pulmonary auscultation is unremarkable. The resting electrocardiogram (ECG) highlights an appearance of incomplete right bundle branch block with the presence of the epsilon wave (Figure 1). A 24-hour Holter ECG is performed highlighting episodes of non-sustained VT.
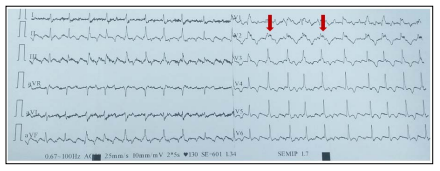
Figure 1: ECG abnormalities in favor of DAVD. Epsilon waves (red arrow); Microvoltage witness to an advanced DAVD; Inversion of the T wave beyond V2
All these elements plead in favor of a ventricular tachycardia whose origin would be right ventricular.
Transthoracic cardiac echocardiography shows a dilated right ventricle (RV), with very impaired systolic function, globally hypokinetic with the presence of aneurysm foci at the level of the apex. It is not hypertrophied, but is the seat of many trabeculations. The surface shortening fraction of the RV is estimated at 25%. The left ventricle is structurally normal.
Cardiac MRI reveals dilation of the right ventricle, with the presence of very extensive intramyocardial fibrosis at the level of the free wall of the RV (Figure 2, Figure 3). It is the site of foci of aneurysms in the subtricuspid (Figure 4), at the level of the pulmonary infundibulum, the tip of the RV, as well as foci of bulging of the anterior and lateral wall of the RV. The left ventricle is non-dilated; with moderately impaired systolic function (LVEF 45%). Coronary, angiography shows a coronary network free of stenosis.
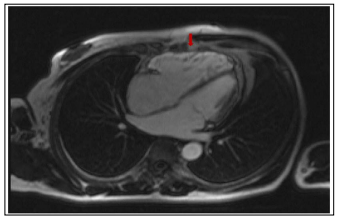
Figure 2: Cine sequence in Steady Stage Free Precession (SSFP), 4-cavity incidence, post injection of contrast product (PDC). RV free wall hypersignal related to fatty infiltration (red arrow)
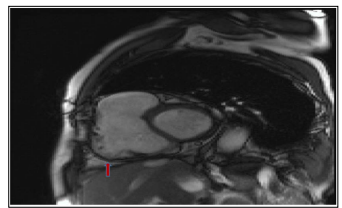
Figure 3: Late enhancement sequence, 4-cavity incidence, contrast enhancement of the RV side of the SIV and of the free wall of the RV related to the fibrosis (red arrow)
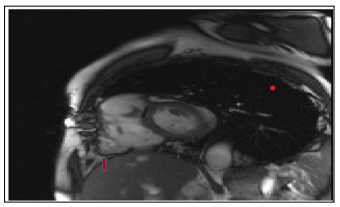
Figure 4: SSFP cine sequence, short axis incidence, presence of an aneurysm in the paratricuspid region (red arrow)
The diagnosis of DAVD is retained. The patient is put on amiodarone, beta-blocker, and ACE inhibitor, loop diuretics, and potassium-sparing agents. Given the family history of sudden death, the patient was fitted with a single-chamber implantable cardioverter defibrillator without incident (Figure 5). A genetic investigation was carried out, without discovery of minor or major signs of DAVD in the collaterals. After one-year follow-up, no rhythmic event was detected.
Figure 5: Single-chamber implantable cardioverter defibrillator (red arrow)
In 1994, the World Health Organization (WHO) classified AVDD as a non-ischemic cardiomyopathy [4]. It is more common in men than in women (ratio 3/1). It is an important cause of sudden death in young athletes, representing less than 10% of sudden death in people under 65 [5]. The clinical picture generally appears between the second and fourth decades of life. Patients may report palpitations secondary to ventricular extrasystoles, ventricular tachycardia, signs of heart failure, syncope, and even sudden death. AVDD is often underdiagnosed [6]. The transmission of the disease is almost always autosomal dominant, which means a 50% risk of transmission to each first degree relative. In very rare cases, corresponding to a syndromic form with cardiac, cutaneous (palmoplantar hyperkeratosis) and appendages (woolly hair) involvement, it is a recessive transmission (Naxos or Carvajal syndrome) [7]. The diagnosis of DAVD is based on a range of arguments. The diagnostic criteria for AVDD were specified in 1994 by the International Task Force of Cardiomyopathies and updated in 2010. A scoring system was established, with major (2 points) and minor (1 point) criteria, taking into account ECG, para-clinical, personal and family parameters. The diagnosis is considered certain in the presence of a score ?4, as probable if the score is 2 of 3 and as possible if the score is 2. Nevertheless, these diagnostic criteria have limitations due to the phenotypic heterogeneity of the DVDA and incomplete penetrance. The diagnosis can be as difficult in the forms beginners or rough, even though there may be a rhythmic risk [8]. Electrocardiogram abnormalities are found in more than 90% of patients with AVDD. About 40% of patients may have a normal ECG, but all will show abnormalities over 6 years, repolarization disorders were common. Delayed terminal activation and fragmented QRS are also prevalent. Low amplitude and QRS fragmentation are independent risk factors for adverse events. The prevalence of epsilon was up to 30%, being one of the major criteria for the diagnosis of DAVD, but it has a low specificity [6, 9]. Diagnosis of this clinical entity has been made easier thanks in particular to advances in imaging. It is commonly established that ultrasound, coupled with cardiac MRI, improves sensitivity and increases it from 75 to around 85%. RV angioscintigraphy, little practiced under our skies, remains a reference examination for the search for small foci of dysplasia, undetectable on MRI. It should be noted that the polymorphism of this disease and the relative initial latency of this disease does not generally allow an early diagnosis, rarely made before the age of 20, and sometimes even at autopsy in the aftermath of a death. Sudden, in athletic adults or not. On the other hand, the discovery of this disease beyond the age of 60 is relatively rare.
The treatment of AVDD is based on the antiarrhythmic component. The efficacy of these treatments is comparable and the data in the literature argue in favor of the absence of a net reduction in mortality compared to the defibrillator [10]. The extension of dysplasia to the left ventricle, the occurrence of resuscitated sudden death or in a first-degree relative are poor prognosis criteria and each constitute a legitimate indication for the implantation of an implantable automatic defibrillator [11 ,12]. Radiofrequency ablation may be attempted in some who have the same form of VT that is not controllable by medical treatment alone, or that causes multiple defibrillation shocks that may cause premature exhaustion of the defibrillator [13, 14]. The appearance of signs of congestive heart failure is considered an evolutionary turning point for this disease. Apart from the usual treatment of heart failure, which is based on ACE inhibitors, beta-blockers, loop diuretics and potassium-sparing agents, the only effective treatment would be heart transplantation [15]. In the meantime, a defibrillator can be implanted.
Finally, genetic investigation is an integral part of overall medical care, the aim of which is to detect the first signs of the disease and to reduce the risk of sudden death linked to rhythmic complications, which currently remains close to 2%/ year without treatment and 0.7%/year with antiarrhythmic treatment. In the future, gene therapy could provide a radical solution for the treatment of diseased desmosomes, and could by restoring normal cellular homeostasis regress or stabilize the disease [16].
Thanks to scientific and technological advances, AVDD has become an easier entity to diagnose and treat. The Task Force criteria, updated in 2010, increase diagnostic sensitivity. The treatment of AVDD has been revolutionized by defibrillator technology. On the other hand, acting directly on the ultra-structural anomaly, which would be the ideal curative treatment, requires even more effort in order to prevent the occurrence of this disease, to stabilize it, and to prevent its complications.