Author(s): Opeyemi Oluwasanmi Adeloye*, Oyeneyin Babatunde David and Olukoju Idowu
Alzheimer disease (AD) is the most common neurodegenerative disease and form of dementia. The peptide amyloid-β (Aβ) is a most therapeutic target in AD on the basis of pathological and genetic suffices that supports a role for this molecule in the disease process. Studies show that Aβ immunotherapies (Active and passive) have been revealed to alleviate cerebral Aβ levels and improve cognition in animal models of AD. In humans, clinical trial phase 2 AN1792 conducted by Elan et al stated that Aβ vaccine was stopped when ~6% of the immunized patients developed meningoencephalitis. However, some plaque clearance and modest clinical improvements were observed in patients following immunization. In this study, Aβ immunotherapies will be discussed. Passive and active method of treatment in human and non-human primate with AD will also be review. Preclinical studies and the limited data from clinical trials and non-human primates’ evidence suggest Aβ immunotherapy as the most effective in preventing or slowing the progression of AD when patients are immunized before or in the very earliest stages of disease onset. AD Biomarkers and imaging technology have improved greatly over the past 11 years and, in the future, can be used to identify pre-symptomatic, at-risk individuals who might benefit from Aβ immunotherapy.
Alzheimer’s disease (AD) is the most common neurodegenerative disease that affects more than 18 million elderly people worldwide. Its prevalence increases with aging, affecting 8-11% of individuals over age 63, and about 42% of persons over 80 years of age. AD is characterized by cognitive dysfunction, memory loss, behavior changes, impairments in the activities of daily living (ADL), incontinent and reduces the quality of life. Studies revealed the neuropathological hallmarks of AD are cerebral amyloid angiopathy and extracellular neuritic plaques formed by Aβ deposits, and intracellular neurofibrillary tangles (NFT) composed of paired helical filaments of hyperphosphorylated protein tau, neuronal loss, neuritic dystrophy, inflammation and gliosis. The aetiology of AD are unknown, but accumulating studies supports the “Aβ hypothesis”, which characterized by insufficient clearance, overproduction or aggregation of Aβ peptide results in neuronal loss and dysfunction underlying dementia in AD.
Evidence from Town. T et al shows that Aβ, a 39-42 residue peptide weighing ~ 4 KD, is formed through the “amyloidogenic pathway” in which amyloid precursor protein (APP) is sequentially cleaved by β- and γ-secretase as opposed to the constituitive non-amyloidogenic pathway that involves processing APP by α-secretase. Mutations in presenilin (PS) 1 and 2 genes can cause onset forms of AD, providing genetic support for the role of Aβ in AD. Researcher findings stated that Apolipoprotein E, especially its Σ4 isoform, α1-antichymotrypsin, and C1q complement factor can greatly increase the aggregation of Aβ. Town. T et al further stated that once Aβ aggregates, its conformational change is thought to initiate a neurodegenerative cascade including impairment of long-term potentiation, changes in synaptic function, and accelerated formation of neurofibrillary tangles (NFT) that will ultimately lead to synaptic failure and neuronal death [1]. Thus, the Aβ cascade has become a central therapeutic target and reducing the Aβ burden in the brain by immunotherapy has developed as a promising strategy for the treatment of AD.
Alzheimer disease (AD) treatments at present do little to modify the disease progression and also provide modest symptomatic benefit for some patients. Results from Humans from clinical trial and non-human primates, active and passive Aβ immunotherapies have become potentially useful disease-modifying strategies for combating AD. Active immunization described administration of synthetic Aβ peptide or Aβ fragments conjugated to a carrier protein and adjuvant to stimulate cellular and humoral immune responses in the host that, in turn, result in the generation of anti-Aβ antibodies. In passive immunotherapy, Aβ-specific antibodies are directly injected into the body of the host, bypassing the need for engagement of the immune system. In both active and passive Aβ immunotherapies, anti-Aβ antibodies remove the Aβ from brain.
Evidence from Schenk et al reported the beneficial effect of Aβ immunotherapy in a preclinical study of Aβ 1-42 active immunization in transgenic (PDAPP) mice. Immunizing mice prior to the onset of pathology reduced levels of cerebral amyloid and produced high serum antibody titers. Also, amyloid deposition was reduced in mice that were immunized after they had developed significant amyloid pathology. This work was later confirmed by active intranasal immunization using a mixture of Aβ1-40 and Aβ1-42 peptides without adjuvant in PDAPP transgenic mice. Two additional studies demonstrated that Aβ vaccination in transgenic CRND8 or APP/PS1 mice strongly improved behavioral performance in learning and memory tasks. Subsequently, different research has confirmed the Aβ-lowering effect of Aβ vaccination in AD-like transgenic mouse models. Illustration of the effect of Aβ immunotherapy on plaque deposition demonstrated below. We intranasally immunized 1 month-old J20 hAPP tg mice with full-length Aβ1-40/42 and an adjuvant, E. coli heat labile enterotoxin LT(R192G), for 11 months. Abundant plaque deposition was seen in hippocampus and cortex of untreated, agematched control J20 mice however, Aβimmunized J20 mice had almost no plaque deposition. Small punctate dots of Aβ immunoreactivity remained, often adjacent to blood vessels, possibly indicating clearance. It is clear from this and many other studies that immunizing APP transgenic mice prior to plaque deposition strongly prevents plaque deposition.
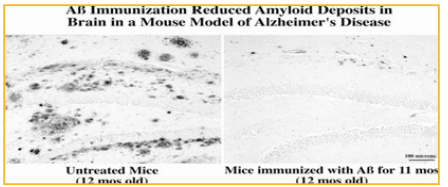
Figure 1: Immunization with full-length Aβ dramatically reduced cerebral Aβ plaque burden in J20 hAPP transgenic mice, a mouse model of Alzheimer’s disease.
In this review, 1 mo-old mice were primed by giving an intraperitoneal injection of 100 μg Aβ1-40/42 synthetic peptide plus 50 μg Complete Freund’s adjuvant. The mice were then boosted weekly by intranasal application of 100 μg Aβ1-40/42 plus 5 μg adjuvant LT (R192G) for a total of 11 months and euthanized at 12 months, an age in which these mice typically accumulate many plaques in cortex and hippocampus (left panel). Immunohistochemical analysis with an Aβ-specific polyclonal antibody R1282. Dennis Selkoe et al revealed a significant reduction in plaque burden in cortex and hippocampus (shown in right panel). Scale bar: 100 μm [2]. [Adopted with permission from Lemere, C.A., Maier, M., Jiang, L., Peng, Y., Seabrook, T.J. Amyloidbeta immunotherapy for the prevention and treatment of Alzheimer’s disease: Lesson from mice, monkeys and men. Rejuvenation Research 9:77-84, 2006.
Different studies about passive immunization studies using Aβ antibodies against the N-terminus, mid-domain, and C-terminus of Aβ have been used in transgenic mice with ADlike pathology. Bard et.al performed passive immunization in PDAPP mice using several different monoclonal anti-Aβ antibodies that targeted various Aβ epitopes and represented different IgG isotypes [3]. The Aβ antibodies were able to enter the central nervous system (CNS), bind plaques and induce clearance of pre-existing amyloid. Later, the authors also stated that antibodies against the N-terminus of Aβ (3D6 against Aβ 1-5 or 10D5 against Aβ sub3-7) were the most effective at reducing brain amyloid. Passive immunization of PDAPP transgenic mice with the 10D5 antibody led to reduced plaque burden, increased peripheral Aβ, improved hippocampal long-term potentiation (LTP), and improved cognitive performance and quality of life. Another monoclonal Aβ antibody, BAM-10 (Aβ 1-12 ), reversed memory impairment in APP transgenic mice, even in the absence of significant amyloid reduction.
Microhemorrhage has been studied following passive immunization with N-terminal Aβ antibodies in APP transgenic mice. In contrast, passive immunization with m266, a centraldomain Aβ monoclonal antibody, did not increase microhemorrhage in mouse brains, although it significantly decreased Aβ plaque pathology and improved cognition. In addition, passive immunization with C-terminal Aβ antibodies has been reported. Bard et al first reported that the 16C11 antibody (against Aβ 33-42 ) failed to lower plaque burden or improve cognitive deficits [4]. Wilcock et al found that transgenic 2576 transgenic mice that were immunized with 2286, an IgG1 C-terminal Aβ antibody against Aβ 28-40, for 3 months showed an improvement in alternation performance in the Y maze, a reduction in both diffuse and compact amyloid deposits, and transient but significant microglial activation. However, this same C-terminal antibody led to a significant increase of CAA-associated microhemorrhage in immunized mice. Subsequently, an IgG2b C-terminal antibody (2H6) and its de-glycosylated version (de-2H6) were shown to reduce Aβ pathology and significantly improve performance in a radial arm water maze. Vascular amyloid and microhemorrhages were reduced in de-2H6-vaccinated mice, possibly because deglycosylation of the antibody decreased its affinity for the Fcγ receptor.
Using APP transgenic mouse models for the study of Aβ immunotherapy has the limitation that the immune response elicited is directed to transgene-expressed human Aβ but not endogenous mouse Aβ protein in brain. Therefore, a preclinical model that is genetically similar to humans, exhibits Aβ pathology with normal aging, and has a comparable immune response, would be of benefit for testing the safety and efficacy of an Aβ vaccine before transitioning to human clinical trials. Rhesus monkeys species, a form of non-human primates such as Macaca mulatta and Caribbean vervets exhibit age-related Aβ deposition similar to AD in humans. Pilot study of vervets of Aβ vaccination in 6 aged demonstrated that active immunization with aged Aβ 1-40/42 over ten months produced appreciable titers of plasma Aβ antibodies that recognized plaques in human, vervet, and APP transgenic mouse brains. Anti-Aβ titers were approximately 1000-fold lower in cerebrospinal fluid (CSF) compared to titers in plasma. In addition, A Aβ protein levels in plasma were elevated while those in CSF and brain tissues were reduced in vaccinated vervets. Inflammation and T cells were absent in the brain tissues of immunized vervets.
Another pilot study revealed in rhesus monkeys using a similar active vaccine with aggregated Aβ 1-42 or aggregated islet amyloid polypeptide (IAPP) and CFA was investigated. Rhesus monkeys that received aggregated Aβ1-42 generated moderate anti-Aβ titers as well as a 5-p10 fold increase of Aβ levels in plasma, as compared with the rhesus monkeys that were vaccinated with aggregated IAPP. In contrast to previous vaccination studies in vervets, cerebral Aβ levels in these younger monkeys was not reduced even though plasma Aβ levels were elevated after vaccination possibly, because they had not reached a plaque-bearing age. Together, these two studies demonstrate that non-human primates display natural Aβ deposition with aging and generated anti-Aβ antibodies when actively vaccinated with an Aβ peptide.
Elan/Wyeth in 2000 and 2001 carried out extensive preclinical studies of active immunization with pre-aggregated Aβ showed promising benefits such as stimulating high antiAβ antibody titers in plasma, and consequently reducing cerebral Aβ burden, as well as preventing or reversing cognitive decline in different mouse models. After a singledose Phase I study in 24 patients and a multiple-dose Phase I study in 80 patients with mild to moderate AD in the United Kingdom, the immunization of Aβ1-42 peptide (AN1792) in combination with the adjuvant QS-21 showed good tolerability in patients and, 58% of the patients with multipledose vaccination developed an anti-Aβ humoral response. Elan/Wyeth further stated that there were few adverse effects and some evidence of possible improvement in one of the clinical measurements. Next, a Phase IIa study was initiated in which 372 mild to moderate AD patients were enrolled to receive either AN1792 (300 patients) or placebo (72 patients). The trial was stopped due to the development of meningoencephalitis in 18 of the 300 (6%) immunized AD patients. Although the adverse events correspond not to antibody response, it has been suggested that they may have been initiated by activation of cytotoxic T cells and/or autoimmune reactions. Subsequent reports showed that even brief active immunization with AN1792 succeeded to some degree in generating an anti-Aβ antibody response, clearing Aβ from brain, and modestly stabilizing cognitive function. However, Holmes and colleagues reported that small group of immunized AD patients who came to autopsy several years after the trial was stopped showed no significant differences between placebo and AN1792 vaccinated groups in survival outcomes or time to severe dementia, or in cognitive measures, even though large areas of cortex were devoid of amyloid plaques. It is quite possible that the disease process, including synaptic and neuronal loss, was too far along when these AD patients entered the trial, suggesting that Aβ vaccination may have its best effects if given prior to or in the very early stages of AD. Active Aβ vaccine trials are current underway, sponsored by pharmaceutical companies such as ELAN/Wyeth, Novartis, Merck, Affiris, and United Biomedical.
Direct injection of anti-Aβ antibodies may be an easy and relatively safe method to provide Aβ antibodies without eliciting Th1-mediated autoimmunity. Intravenous immunoglobulin (IVIg) antibodies, a pooled mixture of human antibodies including anti-Aβ antibodies, were found to interfere with the oligomerization and fibrillization of Aβ peptide protect neurons exposed to toxic concentrations of Aβ peptide and promote the clearance of Aβ peptide from the brain, suggesting that IVIg treatment may be useful in humans as a type of passive immunotherapy to treat AD. In a small pilot study IVIg reduced Aβ peptide levels in the CSF and significantly increased the Aβ levels in blood, suggesting that the antibody mixture induced efflux of Aβ from the brain to the periphery. Furthermore, some improvement in cognitive function was seen in immunized patients in the absence of adverse events. Anti-Aβ antibodies were detected in CSF of the patients subsequent to IVIg treatment, indicating that IVIg antibodies may cross the blood- brain barrier (BBB) and thereby affect Aβ levels in the brain. Several other clinical trials have shown evidence of the potential of IVIg immunotherapy for AD but these trials were carried out only in only a small number of AD patients. More recently, larger phase III clinical IVIg trial in AD patients was initiated by Baxter Biosciences and the Alzheimer’s Disease Cooperative Study (ADCS). In addition, other passive Aβ vaccine clinical trials are underway, sponsored by pharmaceutical companies such as ELAN/Wyeth/Janssen, Eli Lilly, Pfizer, HoffmanLaRoche, Genentech, and GlaxoSmithKline.
The use of N-terminal Aβ derivatives as immunogens was hypothesized to generate a strong humoral immune response while avoiding a deleterious Aβ-specific T cell response because the dominant B cell epitope for anti-Aβ antibodies generated by active immunization with full-length Aβ was found to reside within the first 15 amino acids of the Aβ N-terminus, which was later refined to Aβ1-5, 1-7, 1-8, 1-9, 1-11, 1-16, 4-10, and Ab3-7 in mice, Aβ 1-7 in vervets, and Aβ1-18 in humans while dominant Aβ T cell epitopes have been mapped to Aβ6-28 or beyond Aβ1-15 in mice and Aβ16-33 in humans. This hypothesis has been confirmed by several groups using short Aβ fragment active vaccines. AD trangenic mice immunized with K6Aβ1-30-NH2 vaccine containing 6 lysines linked to the first 30 residues of Aβ, K6Aβ1-30[E18E19] vaccine with mutations at positions 18 and 19, PADRE-Aβ1-15 epitope vaccine containing the Aβ1- 15 in tandem with the synthetic pan HLADR-binding peptide (PADRE) or another epitope vaccine composed of two copies of Aβ1-11 fused with PADRE resulted in generation of antiAβ antibodies, reduction in plaque burden and/or cognitive impairment, and reduction or abolishment of autoreactive T cell responses. We found that while Aβ1-15 peptide was less immunogenic than Aβ1-40/42 for antibody production in wild-type mice intranasal boosting with dendrimeric Aβ1- 15 (16 copies of Aβ1-15 on a lysine tree; dAβ1-15) and a mucosal adjuvant after a single priming injection of Aβ1- 40/42 in J20 hAPP mice resulted in high anti-Aβ antibody titers and reduced cerebral amyloid plaque burden. Later, we found that intranasal delivery of dAβ1-15 with LT(R192G) without a priming injection, also induced robust anti-Aβ titers and lowered cerebral Aβ levels and plaques in the brains of J20 APP tg mice without inducing an Aβ-specific cellular immune response. Intranasal delivery of 2 short Aβ immunogens, 2xAβ1-15 and R2xAβ1-15 (tandem repeat of Aβ1-15 linked by 2 lysine residues, without or with an RGD motif at the N-terminus, respectively) resulted in high antiAβ antibody titers without an Aβ-specific T cell response, and reduced plaque load. In the case of the 2xAβ1-15 vaccine, improved memory acquisition in the Morris water maze in J20 APP tg mice was observed. Numerous other short Aβ fragment active vaccines are currently under investigation.
DNA vaccination may have potential as a treatment for AD because it is simple, easily modified, and may not require the use of an adjuvant. Immune responses of the host can be easily manipulated to obtain a Th2-type reaction. Initially, Aβ DNA vaccines were produced using adeno-associated virus vectors or adenovirus vector. Recently, researchers have focused on developing non-viral plasmid vectors because such DNA vaccines can be mass-produced at a low cost and have no possibility of viral infection or transformation.
Studies suggests Aβ1-42 DNA vaccination with or without adjuvants has been shown to be efficient for breaking host Aβ1-42 tolerance and inducing a Th2 immune response significantly reducing the cerebral Aβ burden in different AD-like transgenic mouse models and reducing CAA, high-molecular-weight oligomers, and Aβ trimers in TgCRND8 mice. Movsesyan et al investigated a shorter DNA epitope vaccine containing three copies of Aβ1-11 fused with a foreign T helper epitope (PADRE), and linked to macrophage-derived chemokine (MDC/CCL22) or three copies of Complement 3d (3C3d) for adjuvant activity. Vaccination of 3xtrangenic-AD mice (encoding mutant human APP, PS1 and Tau) with the pMDC-3Aβ1-11-PRE construct led to high titers of Th2-biased anti-Aβ antibodies. Aβ pathology and glial activation were diminished in the absence of microhemorrhage and cognitive deficits were improved. The 3Aβ1-11-PADRE-3C3d construct resulted in similar beneficial effects on antibody response and Aβ burden.
Okura studies revealed the immunized APP23 transgenic mice with non-viral Aβ DNA vaccines prior to Aβ deposition (prevention) or after the onset of Aβ deposition (therapy) in the brain. Cerebral Aβ burden was reduced in immunized mice following the prevention trial. Cerebral Aβ burden was reduced ~ 50% by the age of 18 months in the therapeutic trial. Neuroinflammation and T cell responses to Aβ peptide were absent in immunized APP23 and wildtype mice, even after long-term vaccination.
Another strategy to increase the generation of Aβ antibodies and the safety of active immunization is to optimize administration routes of the vaccine delivery. For example, we demonstrated that intranasal administration of Aβ peptides, in the absence of adjuvant, induced a modest anti-Aβ antibody response sufficient enough to significantly reduce cerebral Aβ levels in PDAPP mice. Later, we found that adjuvant, E. coli heat labile enterotoxin LT (R192G), significantly enhanced anti-Aβ antibody generation in wildtype and APP tg mice when short Aβ peptide immunogens, dAβ1-15 [66] and 2xAβ1-15 were delivered intranasally. In both studies, intranasal Aβ immunization using LT (R192G) led to a predominantly Th2-biased immune response and a lowering of cerebral Aβ in the absence of any adverse effects.
Transcutaneous immunization utilizes antigen presentation by Langerhans cells in the skin. Previously, we reported that transcutaneous immunization with dAβ1-15, but not Aβ1-40/42, and the adjuvant LT (R192G) resulted in moderate Th2-biased anti-Aβ antibody titers in wildtype mice. Subsequently, Town and colleagues showed that transcutaneous immunization with Aβ1-42 with cholera toxin adjuvant resulted in robust anti-Aβ antibody titers, reduced cerebral Aβ levels, and increased Aβ in blood, while avoiding T cell infiltration into brain and cerebral microhemorrhage.
An oral DNA vaccine consisting of an adeno-associated viral vector carrying Aβ cDNA (AAV/Aβ) without adjuvant induced the expression and secretion of Aβ1-43 or Aβ1-21 in transgenic 2576 mice, leading to the generation of longlasting anti-Aβ antibodies. A single oral administration of AAV/Aβ to 10 month-old transgenic 2576 mice protected against progressive cognitive impairment and decreased Aβ deposition, insoluble Aβ, soluble Aβ oligomers (Aβ56), microgliosis, and synaptic degeneration. Oral immunization of Aβ peptide-loaded microparticles also induced long-term anti-Aβ antibody production in female BALB/c mice.
Janus C, et al studies revealed the second-generation Aβ vaccines have been tested in several non-human primate species [5]. Non-human primates develop Aβ plaque deposition with aging, although the degree of deposition varies among species. Administration of site-specific UBITh Aβ vaccine (Aβ1-14) in baboons and macaques showed that N-terminal-directed anti-Aβ antibodies were generated while bypassing any adverse Aβ cellular immune responses. The vaccine showed significant efficacy in both non-human primate species, suggesting a potential versatility of the vaccine. Furthermore, anti-Aβ antibodies generated by vaccinated monkeys sequestered toxic Aβ from the CNS into the periphery. In addition, repeat dosing with the UBITh vaccine did not appear to be detrimental or toxic in macaques, indicating it was a safe and well-tolerated vaccine in adult macaques. Another recent evidence active Aβ vaccination study in lemurs showed that moderate to robust anti-Aβ IgM and IgG titers were generated in those lemurs immunized with several Aβ derivatives described above, K6Aβ1-30 and K6Aβ1-30[E18E19], with alum adjuvant. An increase of Aβ1-40 in plasma suggested that resulting antiAβ antibodies may bind to Aβ in the brain and draw it to the periphery.
Similar to non-human primates, the Aβ gene in dogs is similar to human Aβ and aged canines, in particular, beagles, accumulate Aβ plaques in the brain with normal aging. Head and colleagues immunized aged beagles presumed to have cerebral Aβ deposition, with fibrillar Aβ1-42 and alum. Aβ-vaccinated beagles generated strong anti-Aβ antibody responses and had reduced Aβ plaque load in several brain regions. However, Aβ vaccination in aged beagles did not ameliorate cognitive impairments, suggesting that early vaccination may be needed to see cognitive benefits.
Town T, et al. demonstrated passive Aβ immunization has been shown to have some beneficial effects on synaptic plasticity and neuronal function. Chauhan et al. also reported that intracerebroventricular (i.c.v.) infusion of Aβ antibodies protected APP transgenic mice from synaptic loss and gliosis [6]. Klyubin and co-workers reported that i.c.v. infusion of 4G8, a monoclonal antibody directed to the mid-region of Aβ (Aβ17-24), prevented synaptic plasticity disruption induced by naturallyoccurring, cell-derived Aβ oligomers, and by human CSF containing Aβ dimers. A study by SpiresJones et al. found that immunization with the Aβ monoclonal antibody 3D6 that recognizes the free N-terminus enhanced structural plasticity in PDAPP mouse brain.
Recent evidence suggests that Aβ oligomers (dimers, trimers, tetramers, etc.) rather than monomers or fibrils may be the major toxic agents that specifically inhibit LTP and synaptic plasticity in AD. As a result, there is a surge of passive Aβ immunotherapies focusing on inhibiting or reversing Aβ oligomerization using specific anti-oligomer antibodies. One example of an oligomer conformation-specific antibody, NAB61, was used to immunize aged transgenic 2576 mice and it showed significant improvement in spatial learning and memory, without altering brain amyloid deposition or APP processing. This result supports the hypothesis that cognitive deficits in APP tg mice are at least partially caused by toxic soluble Aβ oligomers.
Interestingly, Britschgi et al. recently reported that abundant natural, endogenous anti-Aβ antibodies (IgGs), especially those reactive against high molecular mass assemblies of oligomeric Aβ and pyroglutamate or oxidized residues of Aβ, can be found in plasma and CSF of both AD patients and healthy controls. Once isolated from human blood, these natural antibodies protected primary neurons from oligomeric Aβ toxicity in vitro. Plasma IgG reactivities against several Aβ forms, including oligomeric Aβ1-42 in particular, as well as amyloidogenic non-Aβ peptides were reduced with aging and AD in humans. In addition, natural Aβ IgGs observed in plasma samples from our aged vervets were similar to those in human AD patients. Active immunization with Aβ1-40/42 in aged vervets led to high titers not only against conformation-specific Aβ assemblies, but also against non-amyloidogenic peptides. Thus, it appears that enhancing the generation of neuroprotective natural anti-Aβ antibodies or passive applying them to the elderly might be beneficial for the prevention and treatment of AD.
Arbel et al. reported a novel approach to inhibit Aβ production via antibodies against the beta-secretase cleavage site of the APP. Long-term systemic administration of this anti-beta-site antibody in Tg2576 mice improved cognitive deficits, reduced inflammation, and decreased the incidence of microhemorrhage without inducing any peripheral autoimmunity responses. However, cerebral Aβ levels were unchanged by the antibody treatment.
Alternative strategies to improve the efficacy of passive immunization and reduce its side effects have been reported. Compared with systemic immunization of Aβ Mab 6E10, prolonged i.c.v. infusions by osmotic mini-pump dosedependently reduced the parenchymal plaque burden, astrogliosis, and dystrophic neurites at much lower doses. In addition, side effects observed after administration of someN-terminal Aβ antibodies such as microhemorrhage were reduced by modulating antibody dose deglycosylating whole IgG Aβ antibody removing the Fc portion by proteolysis to produce Fab’2 or designing recombinant Fab or single-chain variable fragment.
Studies shows the mechanisms by which Aβ is cleared from the brain via active or passive vaccination are not clear yet, however, several major hypotheses have been proposed including microglia-mediated phagocytosis, antibodymediated alterations of Aβ aggregation and neutralization of Aβ toxicity, peripheral sink, and intracerebral sequestration of Aβ in a monomeric state.
Intracellular Aβ immunoreactivity within microglia and macrophage in PDAPP tg mice immunized with Aβ monoclonal antibodies indicated that peripherally administered Aβ antibodies can cross the BBB, bind to Aβ in plaques and trigger the Fc receptors (FcR)-mediated microglial phagocytosis. The mechanism of microglial FcRdependent Aβ clearance was further proven in an ex vivo assay with sections of PDAPP tg or AD brain tissue. The data showed that antibodies against Aβ peptide activated microglial cells and subsequently removed plaques via Fc receptor (FcR)- mediated phagocytosis. Active vaccination with Aβ1-42 in PDAPP tg mice and in AD patients also showed evidence that the reduction of compact and densely stained Aβ deposits were associated with microglial Fc-receptor phagocytosis. Additionally, the activation of microglial phagocytosis and removal of Aβ deposits were also observed in mice treated with Aβ nonviral DNA vaccines. The combined results of numerous active and passive Aβ immunotherapy studies indicate that FcR-mediated activation of microglia could be a central mechanism of reducing Aβ load in brain.
However, effective clearance of Aβ deposits were also observed in Aβ1-42 immunized FcRγ-/- Tg2576 mice or in APP transgenic mice treated with F(ab’)2 fragments of 3D6 antibody (antibody without Fc portion) implying that the FcR-mediated phagocytosis is not the only mechanism involved in microglia-induced plaque removal. These findings demonstrate that Fc-independent mechanisms, in addition to Fc-dependent mechanisms, are capable of mediating the effects of active and passive Aβ immunization
Certain anti-Aβ antibodies have been reported to directly bind to Aβ and either prevents oligomerization and fibril formation of Aβ or dissolve Aβ aggregates in vitro. Specific conformational Aβ antibodies target existing plaques in the brain and lead to direct disassembly in vivo. These findings suggest that direct interaction of anti-Aβ antibodies with Aβ could potentially affect Aβ aggregation both in vitro and in vivo. In addition, Aβ antibodies that recognize an oligomeric conformation have been shown to reverse or ameliorate the cognitive deficits in vivo, indicating that another possible mechanism of Aβ immunotherapy may be the blockade of neurotoxicity induced by Aβ oligomers.
The peripheral sink mechanism was first proposed by DeMattos and colleagues in the study using m226, an Aβ midregion antibody with a high affinity for soluble Aβ, in PDAPP tg mice. Later, this mechanism was confirmed by active [60, 61, 112, 113] and passive [30, 114-116] Aβ immunization studies. These studies suggest that Aβ antibody in plasma can reduce circulating Aβ level by directly binding to plasma Aβ, which in turn disrupts the brain-blood equilibrium of Aβ and drives the removal of soluble Aβ from the brain. Furthermore, the studies performed by Deane and colleagues indicate that net clearance can involve a peripheral sink effect and an active FcR-mediated process at the BBB as well.
Another mechanism, intracerebral sequestration of monomeric Aβ, has been proposed recently by Yamada and colleagues, who found that peripheral injection of anti-Aβ monoclonal antibody, m266, with high affinity to soluble Aβ, followed by intracerebral microinjection of radiolabeled Aβ1-40 in wildtype mice slowed Aβ clearance from mouse brains. In addition, peripheral administration of m266 antibody in young, pre-plaque APP transgenic mice led to increased levels of monomeric, antibody-bound Aβ in brain without affecting total soluble Aβ levels in brain. Their results indicate that certain Aβ antibodies may sequester soluble, monomeric Aβ in the brain thereby preventing the formation of multimeric Aβ and related neurotoxicity.
McLaurin J, et al stated that antibody-independent immune cell-mediated plaque clearance, effects on Aβmediated vasoconstriction, and modulation of CNS cytokine production as well as IgM-mediated hydrolysis of Aβ may be other possible mechanisms underlying Aβ immunotherapy. Overall, as illustrated in Fig. (2), there may be many mechanisms involved in Aβ clearance and cognitive improvement resulting from active and passive immunization. These mechanisms may act independently, concomitantly or sequentially for Aβ immunotherapy depending on the severity of the disease, antibody specificity and Ig isotype, and specific animal model used.
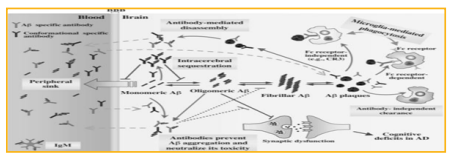
Figure 2: Potential mechanisms underlying Aβ immunotherapy in AD models.
Antibody-mediated microglial FcR-dependent and FcRindependent clearance of plaques by phagocytosis; antibodymediated direct disassembly of plaques; prevention of Aβ aggregation and neutralization of oligomer toxicity; peripheral sink effect by clearance of circulating Aβ; intracerebral sequestration of Aβ in a monomeric state; hydrolysis of Aβ by IgM; and antibody-independent, cell mediated plaque clearance have all proposed to play roles in the removal of Aβ from the brain by Aβ immunotherapy in AD models. These potential mechanisms may act concomitantly or sequentially and play independent roles depending on the stage of AD pathogenesis and type of antibody, as well as the specific animal model used.
Evidence from animal model and human shows effectiveness of Aβ immunotherapy in AD patients. Studies evidence revealed the induce anti-Aβ antibody generation, reduce cerebral Aβ levels, and in some studies, stabilize or improve cognition. Evidence from the AN1792 trial, many groups, both academic and commercial, has generated novel active and passive Aβ vaccines. The goals of passive Aβ immunization are to lower safely cerebral Aβ via monthly injections of antibodies. The goals of active Aβ immunization are to induce long-lasting, safe, and cost-effective vaccines to lower cerebral Aβ and/or prevent Aβ aggregation. The ultimate goal for both types of vaccines is to prevent or diminish the downstream effects of Aβ on synapses and neurons, thereby providing cognitive benefits. Multiple active and passive Aβ immunotherapy clinical trials are currently underway. In addition, tau-related vaccines are also being studied in tanglebearing tau transgenic mouse models. As the average lifespan increases worldwide, the number of AD patients who suffer from this devastating neurodegenerative disease grows as well. Therefore, it is necessary to find a safe and effective way to prevent or cure the disease as soon as possible. Although there is much work to be done, we remain hopeful that Aβ immunotherapy, either alone or in combination with other therapies, will succeed in preventing or treating AD [7-64].